Comprehensive understanding of bacteria could lead to new insights into many organisms
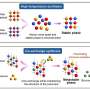
 enabled both a metabolic reconstruction (left) and an atomic-level structure determination/modeling of T. maritima proteins (right). Integration of these two approaches enabled detailed information to be acquired for every reaction in the network (top).")
(PhysOrg.com) -- Investigators at Burnham Institute for Medical Research, University of California, San Diego, The Scripps Research Institute, Genomics Institute of the Novartis Research Foundation and other institutions have constructed a complete model, including three dimensional protein structures, of the central metabolic network of the bacterium Thermotoga maritima (T. maritima).
This is the first time scientists have developed such a comprehensive model of a metabolic network overlaid with an atomic resolution of network proteins. The analysis of the model, among others, highlights the important role of a small number of essential protein shapes, lending new insights into the evolution of protein networks and the functions within these networks. The study was published in the journal Science on September 18.
Combining biochemical studies, structural genomics and computer modeling, the researchers deciphered the shapes, functions and interactions of 478 proteins that make up T. maritima's central metabolism. The team also found connections between these proteins and 503 unique metabolites in 562 intracellular and 83 extracellular metabolic reactions.
"We have built an actual three dimensional model of every protein in the central metabolic system," said Adam Godzik, Ph.D., director of Burnham's Bioinformatics and Systems Biology program. "We got the whole thing. This is analogous to sequencing an entire genome."
With this data, scientists can simulate metabolism simultaneously on a biochemical and molecular level. This information has the promise to expand computer modeling to allow investigators to simulate the interactions between proteins and various compounds in an entire system. Furthermore, the procedure developed in this study could be applied to study many other organisms, including humans. It could potentially help identify both positive and adverse drug reactions before pre-clinical and clinical trials. The research may also have applications in energy research, as bacteria like T. maritima can be engineered to more efficiently produce hydrogen, a key source of clean energy.
"In addition to the systematic analysis of interacting components, the next challenge is addressing the levels or scales of biological organization, ranging from molecules to an individual and even to populations," said co-author John C. Wooley, Ph.D., of UC San Diego's Center for Research in Biological Systems and the California Institute for Telecommunications and Information Technology. "This work, by including both functional and architectural details, takes that first step and provides a novel, enriched view of the complexity of life."
Researchers were surprised by the degree of structural conservation within the network. Of the 478 proteins, with 714 domains, there were only 182 distinct folds. This supports the hypothesis that nature uses existing shapes, slightly modified, to perform new tasks.
The team used genomic, metabolic, and structural reconstruction to determine the network down to the atomic level. They then classified metabolic reactions based on whether they were similar, connected or unrelated and found that enzymes that catalyze similar reactions have a higher probability of having similar folds. In addition, using a reductive evolution simulation approach, they uncovered the absolutely essential proteins to support a minimal viable network.
"We were able to put together the information from the network biology as well as the protein structural biology," said co-author Bernhard Palsson, Ph.D., a UC San Diego bioengineering professor who leads the Systems Biology Research Group. "This is the first time this has been accomplished. We are in a position to study microorganisms in much greater detail, including those that are important in health care and those that are of environmental concern."
Source: University of California - San Diego (news : web)