Researchers find possible treatment for spinal muscular atrophy
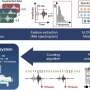
 targeting a specific intronic sequence corrects aberrant splicing of Survival Motor Neuron 2 (SMN2) and restores high levels of functional SMN protein in patient cells of spinal muscular atrophy (SMA). Prominent green dots (see in \"SMN section\") represent well-organized SMN bodies (gems) in the nucleus of the 3UP8-treated cells (bottom two panels). Gem contains another protein ZPR1 (see red dots in \"ZPR1 section\") that co-localizes with SMN protein in the nucleus of the cell (see white dots in \"Merge section\").
The control oligonucleotide (F8) that targets an unrelated sequence had no effect on SMN or ZPR1 levels in patient cells (top two panels). Cell were transfected with the same amount of antisense oligonucleotide (compare the color intensity in \"Oligo section\"). Credit: Courtesy Ravindra Singh")
Spinal Muscular Atrophy is the second-leading cause of infant mortality in the world.
Ravindra Singh, associate professor in biomedical sciences at Iowa State University's College of Veterinary Medicine, would like to see Spinal Muscular Atrophy lose its high ranking and even slide off the list altogether.
Most Spinal Muscular Atrophy sufferers -- more than 95 percent -- have a mutated or deleted gene called Survival Motor Neuron 1 (SMN1) that doesn't correctly do its job of creating functional SMN proteins.
Singh's solution is to replace that poor-performing gene with another gene.
Humans need a certain level of SMN protein to ward off Spinal Muscular Atrophy.
When SMN1 fails to create functioning proteins, Spinal Muscular Atrophy is the result.
There is a gene already in humans that looks very much like SMN1, so much so that it's called SMN2. The SMN2 gene doesn't seem to serve any function that researchers can identify.
Singh has discovered a way of using SMN2 to produce the working SMN protein. When SMN2 makes enough SMN, it compensates for the mutated or malfunctioning SMN1 gene.
All proteins in human bodies are made by copying genes. This copy is called pre-mRNA.
Pre-mRNA then becomes mRNA by splicing out certain parts of the sequence that are non-coding, meaning they don't help the function of the gene.
These non-coding portions of the pre-mRNA are called intronic sequences, sometimes referred to as junk sequence because it is originally copied from junk DNA.
SMN2 normally doesn't produce normal protein because of the presence of a specific intronic sequence in the gene or DNA.
To make SMN2 behave as SMN1, Singh has introduced a small antisense oligonucleotide that blocks this specific intronic sequence.
When the intronic sequence is blocked, SMN2 produces normal proteins and acts, in effect, like SMN1.
"The significance of our work is that we have this stuff called junk DNA in SMN2," said Singh. "We found that we could get SNM2 to behave as SMN1 by introducing a small oligonucleotide. It is a very simple experiment if you think about it."
The resulting proteins are normal just like a regular cell - free from Spinal Muscular Atrophy.
"Our cells are healthy and survive," he said. "From that point of view, this is a major achievement."
Singh, along with his team Natalia Singh and Maria Shishimorova, both of Iowa State University's biomedical services department; Lu Cheng Cao, University of Massachusetts Medical School, Worcester; and Laxman Gangwani, Medical College of Georgia, Augusta, have their research highlighted as the cover story on this month's issue of the journal RNA Biology. Their research (link to research) is the most downloaded story on the RNA Biology page of the Web site Landes Bioscience.
Spinal Muscular Atrophy affects 1 in 6,000 to 1 in 10,000 children born every year. One in 40 people are carriers of the disease -- they don't have the symptoms, but could pass the disease to their children.
Most children born with the most severe type of SMA die within two years.
Using this junk sequence in SMN2 to restore the high levels of functional SMN protein could eliminate Spinal Muscular Atrophy caused by deletion or mutation in SMN1.
Singh believes this technology could also work treating other diseases.
"We know that Parkinson's disease, Alzheimer's disease, cystic fibrosis, multiple sclerosis and cancer all come from genes that are aberrantly spliced," he said.
"If this is a model disease, meaning we succeed in treating Spinal Muscular Atrophy, we will know how to correct splicing of other genes in other diseases," he said.
Source: Iowa State University (news : web)