Protein structures revealed at record pace

Scientists at the U.S. Department of Energy's Lawrence Berkeley National Laboratory have developed a fast and efficient way to determine the structure of proteins, shortening a process that often takes years into a matter of days.
The high-throughput protein pipeline could allow scientists to expedite the development of biofuels, decipher how extremophiles thrive in conditions that kill most organisms, and better understand how proteins carry out life's vital functions.
The technique will help scientists keep pace with the growing flood of data stemming from genomic studies of organisms and environmental samples such as seawater and soil. Every new gene identified in these studies codes for a protein, and the structure of each protein must be characterized in order to determine what it does. Current structural characterization techniques are slow, however, meaning newly discovered proteins and their many complexes keep piling up, their function remaining a mystery.
"There's a bottleneck in structural genomics, and our system addresses that," says Greg Hura, a scientist in Berkeley Lab's Physical Biosciences Division. He developed the technique with John Tainer of Berkeley Lab's Life Sciences Division and the Scripps Research Institute in La Jolla, CA. Michael Adams and other scientists from the University of Georgia also contributed to the research.
Their work is published in the July 20 online edition of the journal Nature Methods.
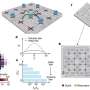
The team developed the protein pipeline at the Advanced Light Source (ALS), a national user facility located at Berkeley Lab that generates intense light for scientific research. At a beamline called SIBYLS, they used a technique called small angle x-ray scattering (SAXS), which can image a protein in its natural state, such as in a solution, and at a spatial resolution of about 10 angstroms, which is small enough to determine a protein's three-dimensional shape. The brilliant light generated by the Advanced Light Source minimizes the amount of material required for each experiment, which makes the technique practical for almost any biomolecule.
To maximize speed, Hura installed a robot that automatically pipettes protein samples into position so they can be analyzed by x-ray scattering. And to analyze the resulting data, they used the supercomputing resources of the U.S. Department of Energy's National Energy Research Scientific Computing Center (NERSC), which is based at Berkeley Lab. The supercomputer's clusters can churn through data for 20 proteins per week, or more than 1000 macromolecules per year.
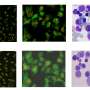
The result is a system that moves at breakneck speed compared to current techniques used to determine the shape and structure of proteins: x-ray crystallography and nuclear magnetic resonance. Recently, in the span of one month, the team used the system to resolve the structure of 40 proteins from Pyrococcus furiosus, a microscopic extremophile that can live at 100°C.
"This would have taken several years with x-ray crystallography," says Hura. "What used to take years, now can takes weeks."
Adds Tainer, "We can now obtain structural information in solution on most samples, rather than the 15 percent obtained by the best of the current Structural Genomics Initiative efforts employing nuclear magnetic resonance and crystallography. "
The Berkeley Lab team chose P. furiosus because it is an intriguing candidate for the production of clean energy and other applications. It has a pathway that produces hydrogen, which is a potential alternative fuel. And many industrial processes are highly acidic and very hot — conditions that P. furiosus loves.
"If we could learn which of the organism's proteins allow it to thrive in these conditions, then maybe we can apply them to energy production and other applications," says Hura.
Future synthetic biology efforts may involve taking a useful protein or a network of proteins from one microbe, and importing it into another microbe. In order to do this, scientists must learn how much of the network needs to be imported and still have it be able to do its job. This requires analyzing individual proteins in hundreds of different conditions.
"This is where our system will have a big impact. We can do this type of structural analysis in a matter of weeks, as opposed to years with crystallography," says Hura.
Of course, such speed doesn't come without tradeoffs. X-ray crystallography yields extremely high-resolution images, while small angle x-ray scattering yields a protein's shape at a much lower resolution of about 10 angstroms (one angstrom is one ten-millionth of a millimeter).
But the level of information offered by x-ray crystallography isn't always necessary. Sometimes, simply knowing if one protein is similar in shape to another is enough to learn its function. And SAXS makes up for what it lacks in precision by providing accurate information on the shape, assembly, and conformational changes of proteins in solution.
"We can have less information and still answer the questions that need to be answered," says Hura, adding that their technique will help usher in the next phase of genomics research. "The number of genes being identified is growing at a huge rate. We need to keep pace with this and learn about all the proteins encoded in these genes."
Adds Tainer, "This pipeline is an example of the stunning impact we can achieve by combining physics and engineering with structural biology, which is possible at government labs like Berkeley Lab."
The multidisciplinary work, which was conducted at Berkeley Lab's Advanced Light Source at beamline 12.3.1, also known as SIBYLS (Structurally Integrated BiologY for Life Sciences), relied on resources provided by three separate offices within the DOE Office of Science (SC). This work itself was supported in part by SC's Office of Biological and Environmental Research (BER). The ALS is supported by SC's Office of Basic Energy Sciences, while the beamline is supported in part by BER. NERSC is funded by SC's Office of Advanced Scientific Computing.
To aid communication of results, the team created a web-accessible database, www.Bioisis.net, which archives all experimental details associated with each analyzed sample.
Source: Lawrence Berkeley National Laboratory