The battle for CRTC2: How obesity increases the risk for diabetes
 and Yiguo Wang are researchers with the Salk Institute for Biological Studies. Credit: Courtesy of the Salk Institute for Biological Studies")
Obesity is probably the most important factor in the development of insulin resistance, but science's understanding of the chain of events is still spotty. Now, researchers at the Salk Institute for Biological Studies have filled in the gap and identified the missing link between the two. Their findings, to be published in the June 21, 2009 advance online edition of the journal Nature, explain how obesity sets the stage for diabetes and why thin people can become insulin-resistant.
The Salk team, led by Marc Montminy, Ph.D., a professor in the Clayton Foundation Laboratories for Peptide Biology, discovered how a condition known as ER (endoplasmic reticulum) stress, which is induced by a high fat diet and is overly activated in obese people, triggers aberrant glucose production in the liver, an important step on the path to insulin resistance.
In healthy people, a "fasting switch" only flips on glucose production when blood glucose levels run low during fasting. "The existence of a second cellular signaling cascade—like an alternate route from A to B—that can modulate glucose production, presents the potential to identify new classes of drugs that might help to lower blood sugar by disrupting this alternative pathway," says Montminy.
It had been well established that obesity promotes insulin resistance through the inappropriate inactivation of a process called gluconeogenesis, where the liver creates glucose for fuel and which ordinarily occurs only in times of fasting. Yet, not all obese people become insulin resistant, and insulin resistance occurs in non-obese individuals, leading Montminy and his colleagues to suspect that fasting-induced glucose production was only half the story.
"When a cell starts to sense stress a red light goes on, which slows down the production of proteins," explains Montminy. "This process, which is known as ER stress response, is abnormally active in livers of obese individuals, where it contributes to the development of hyperglycemia, or high blood glucose levels. We asked whether chronic ER stress in obesity leads to abnormal activation of the fasting switch that normally controls glucose production in the liver." The ER, short for endoplasmic reticulum, is a protein factory within the cell.
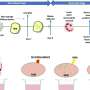
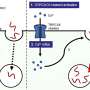
To test this hypothesis the Salk team asked whether ER stress can induce gluconeogenesis in lean mice. Glucose production is turned on by a transcriptional switch called CRTC2, which normally sits outside the nucleus waiting for the signal that allows it to slip inside and do its work. Once in the nucleus, it teams up with a protein called CREB and together they switch on the genes necessary to increase glucose output. In insulin-resistant mice, however, the CRTC2 switch seems to get stuck in the "on" position and the cells start churning out glucose like sugar factories in overdrive.
Surprisingly, when postdoctoral researcher and first author Yiguo Wang, Ph.D., mimicked the conditions of ER stress in mice, CRTC2 moved to the nucleus but failed to activate gluconeogenesis. Instead, it switched on genes important for combating stress and returning cells to health. On closer inspection, Wang found that in this scenario CRTC2 did not bind to CREB but instead joined forces with another factor, called ATF6a.
What's more, like jealous lovers CREB and ATF6a competing for CRTC2's affection—the more ATF6a is bound to CRTC2, the less there is for CREB to bind to. "This clever mechanism ensures that a cell in survival mode automatically shuts down glucose production, thus saving energy," says Wang.
This observation led the researcher to ask what happens to ATF6a following the kind of persistent stress presented by obesity? They found that the levels of ATF6a go down when ER stress is chronically activated, compromising the cells' survival pathway and favoring the glucose production pathway; hyperglycemia wins in conditions of persistent stress.
Explains Wang, "Our study helps to explain why obese people have a stronger tendency to become diabetic. When ER stress signaling is abnormal glucose output is actually increased."
"It is possible that mutations in the highly conserved CRTC2 lead to a predisposition to inappropriate gluconeogenesis," says Montminy, who is now trying to identify natural mutations in CRTC2 that may lead to insulin resistance in carriers.
Source: Salk Institute (news : web)