Researchers improve accuracy of breast cancer prognoses
One of the many unknowns facing women who are diagnosed with breast cancer is predicting the likelihood that the cancer will spread to other parts of the body – metastasize. Researchers from UC San Diego are looking to change that. UCSD bioengineering professor Trey Ideker is pioneering a more accurate approach for predicting the risk of breast cancer metastasis in individual patients.
This work will be published online by the journal Molecular Systems Biology on Tuesday 16 October.
Distant metastases are the main cause of death among breast cancer patients, but physicians have a hard time predicting if a patient’s breast cancer is likely to spread.
The researchers from UCSD and the Korea Advanced Institute of Science and Technology took advantage of new protein interaction databases and identified networks of genes from breast cancer patients – rather than individual genes – that can be used to predict whether a breast cancer tumor is likely to spread.
Their results offer new mechanistic insights into breast cancer metastasis and are more accurate and reproducible than two sets of individual marker genes currently used to help predict the likelihood that a patient’s breast cancer will spread.
“Over the years, large numbers of women have endured unnecessarily harsh treatments, such as aggressive chemotherapy, due to our inability to predict metastasis risks with high accuracy. One of our goals is to improve this situation,” said Trey Ideker, a bioengineering professor from the UCSD Jacobs School of Engineering and the senior author of the new study.
“The next step is to confirm these results in other clinical trials. It will be absolutely crucial to confirm our findings on other patient data before we think too hard about bringing this technology to the clinic,” Ideker said.
The new research may also help researchers discover disease-causing genes and more precisely classify and diagnose cancer and other diseases.
“Our work supports the notion that cancer is a disease of pathways,” said Ideker. “The keys for understanding at least some of these pathways are encoded in protein networks.”
The new study uses the same gene expression data used in two well-known studies: Vijver et al. in Nature and Wang et al. in the Lancet. Each study yielded a set of about 70 single-gene markers that are now used in hospitals to help predict the likelihood of breast cancer metastasis.
“We saw about a 9 percent increase in metastasis prediction accuracy over the two main sets of individual gene markers,” said Ideker, who explained that his team raised metastatic prediction accuracy for breast cancer to roughly 72 percent. “But there is still plenty of room for improvement,” he said.
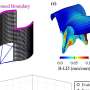
“The big difference between our work and the work outlined in Vijver and Wang is that we painted the existing gene expression data onto newly available maps of protein interactions,” said Ideker. Some refer to these maps as “wiring diagrams.”
By focusing on how the proteins within cells interact, the researchers were able to look at the aggregate behavior of genes that are connected in functional networks. This approach improved their ability to predict which tumors would spread.
Using a mathematical approach for the prediction of metastasis (involving both machine learning and dimensionality reduction), the researchers calculated the average behavior of subnetworks of proteins and used this information to uncover subnetworks that predict metastasis better than individual gene markers.
The team uncovered 149 discriminative subnetworks consisting of 618 genes from the patients from the van de Vijver et al. data set and 243 discriminative subnetworks with 906 genes from the Wang et al. data set.
Each subnetwork is suggestive of a distinct functional pathway or complex, yielding many known and novel pathway hypotheses in organisms for which sufficient protein interaction data have been measured, the authors write in their Molecular Systems Biology paper.
For example, the researchers show that a well-known breast cancer susceptibility gene, P53, plays a central role in several protein subnetworks; it interconnects many expression-responsive genes (genes that show up as potential markers in expression-only analyses). Interestingly, P53 itself does not show up as “significant” in conventional expression clustering or classification methods.
“A key feature of our approach is the ability to identify crucial genes that fly under the radar of conventional gene expression analyses,” said Ideker.
The phenotypic changes most indicative of breast cancer metastasis need not be regulated at the level of expression, the authors write.
The researchers also show that their subnetwork markers are significantly more reproducible between data sets than individual marker genes selected without network information.
Source: University of California - San Diego