May 17, 2016 report
Recent advances in natural product drug design: Chemical fragments and computational methods

(Phys.org)—A review article in Nature Chemistry by Tiago Rodrigues, Daniel Reker, Petra Schneider, and Gisbert Schneider of the Swiss Federal Institute of Technology looks at recent advances in medicinal chemistry including how fragment-based libraries and computational studies have furthered the field of natural product discovery.
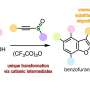
Drug design often involves synthetically mimicking certain biologically active chemicals in naturally-occurring compounds. The process of synthetically making a naturally-occurring compound is time-consuming and expensive because many natural products are too complex to re-construct in a laboratory setting. Furthermore, it is often difficult to know what chemical segments of a natural product scaffold are necessary for efficacy. More recently, high throughput screening libraries have been used to identify pharmacologically active components of natural products that would serve as potential lead compounds.
Traditionally, in looking for natural product pharmacological candidates, researchers will identify compounds that meet certain criteria. For example, they will look for compounds that follow Lipinski's "rule of five." However, these criteria can be too narrow. For example, studies have shown that about 18% of the natural products in the Directory of Natural Products database violate the "rule of five" in at least two criteria.
Medicinal chemists have been exploring other avenues to identify target compounds including fragment-based drug discovery. In contrast to high throughput libraries, fragment-based libraries are low molecular weight (300 Da) and tend to have lower affinities for the target compound. These low molecular weight compounds, however, are often water soluble and easily functionalized to tailor them for drug design.
Fragment-based libraries using sophisticated computational methods can assist in drug design by dissecting complex molecular structures and identifying scaffold parts that are biologically active. This process takes a natural product whose structure is too complex to be synthetically feasible, and dissects the compound layer-by-layer to identify potential simple compounds for exploration. However, as molecular simplicity increases, its efficacy can decrease.
Because biologically active fragments tend to be less effective compared to their more complex naturally-occurring counterparts, chemists can use software that allows for virtual identification of potential ligands or receptors for drug targets as helpful prediction tools. Software programs such as SPiDER look for chemical properties to match ligands and receptors. The authors of this review used SPiDER to take a "target-centric" approach to predicting which natural product fragments might be potential candidates for drug design. One example, archazolid A, whose structure is atypical for small drugs, was found to target certain proteins involved in the recognition and processing of arachidonic acid. This approach, however, is limited to investigating previously studied targets and remains merely a qualitative tool for drug discovery.
Additional computational studies looked at whether natural products are necessarily "better" leads for drug design. As it turns out, natural products tend to have potentially more chemically reactive sites. These compounds may be frequent hitters because they contain chemically reactive components, leading to a false positive for drug design. Therefore, Rodrigues, et al. then studied whether these natural products tend to interact with multiple targets. Even though natural products tend to have more chemically reactive sites, they do not seem to act as 'promiscuous ligands', while synthetic compounds tend to interact with multiple targets.
There has been a resurgence in investigating natural products for drug design. Advances in technology and computational methods applied to natural-product fragments have led to identification of drug targets as wells as a better understanding of generally which chemical features make natural products good candidates for drug design.
More information: Tiago Rodrigues et al. Counting on natural products for drug design, Nature Chemistry (2016). DOI: 10.1038/nchem.2479
Abstract
Natural products and their molecular frameworks have a long tradition as valuable starting points for medicinal chemistry and drug discovery. Recently, there has been a revitalization of interest in the inclusion of these chemotypes in compound collections for screening and achieving selective target modulation. Here we discuss natural-product-inspired drug discovery with a focus on recent advances in the design of synthetically tractable small molecules that mimic nature's chemistry. We highlight the potential of innovative computational tools in processing structurally complex natural products to predict their macromolecular targets and attempt to forecast the role that natural-product-derived fragments and fragment-like natural products will play in next-generation drug discovery.
Journal information: Nature Chemistry
© 2016 Phys.org