Scientists propose an algorithm to study DNA faster and more accurately

A team of scientists from Germany, the United States and Russia, including Dr. Mark Borodovsky, a Chair of the Department of Bioinformatics at MIPT, have proposed an algorithm to automate the process of searching for genes, making it more efficient. The new development combines the advantages of the most advanced tools for working with genomic data. The new method will enable scientists to analyse DNA sequences faster and more accurately and identify the full set of genes in a genome.
Although the paper describing the algorithm only appeared recently in the journal Bioinformatics, which is published by Oxford Journals, the proposed method has already proven to be very popular—the computer software program has been downloaded by more than 1500 different centres and laboratories worldwide. Tests of the algorithm have shown that it is considerably more accurate than other similar algorithms.
The development involves applications of the cross-disciplinary field of bioinformatics. Bioinformatics combines mathematics, statistics and computer science to study biological molecules, such as DNA, RNA and protein structures. DNA, which is fundamentally an information molecule, is even sometimes depicted in computerized form (see Fig. 1) in order to emphasize its role as a molecule of biological memory. Bioinformatics is a very topical subject; every new sequenced genome raises so many additional questions that scientists simply do not have time to answer them all. So automating processes is key to the success of any bioinformatics project, and these algorithms are essential for solving a wide variety of problems.
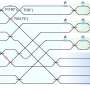
One of the most important areas of bioinformatics is annotating genomes – determining which particular DNA molecules are used to synthesize RNA and proteins (see Fig. 2). These parts – genes – are of great scientific interest. The fact is that in many studies, scientists do not need information about the entire genome (which is around 2 metres long for a single human cell), but about its most informative part – genes. Gene sections are identified by searching for similarities between sequence fragments and known genes, or by detecting consistent patterns of the nucleotide sequence. This process is carried out using predictive algorithms.
Locating gene sections is no easy task, especially in eukaryotic organisms, which includes almost all widely known types of organism, except for bacteria. This is due to the fact that in these cells, the transfer of genetic information is complicated by "gaps" in the coding regions (introns) and because there are no definite indicators to determine whether a region is a coding region or not.

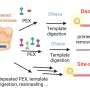
The algorithm proposed by the scientists determines which regions in the DNA are genes and which are not. The scientists used a Markov chain, which is a sequence of random events, the future of which is dependent on past events. The states of the chain in this case are either nucleotides or nucleotide words (k-mers). The algorithm determines the most probable division of a genome into coding and noncoding regions, classifying the genomic fragments in the best possible way according to their ability to encode proteins or RNA. Experimental data obtained from RNA give additional useful information which can be used to train the model used in the algorithm. Certain gene prediction programs can use this data to improve the accuracy of finding genes. However, these algorithms require type-specific training of the model. For the AUGUSTUS software program, for example, which has a high level of accuracy, a training set of genes is needed. This set can be obtained using another program – GeneMark-ET – which is a self-training algorithm. These two algorithms were combined in the BRAKER1 algorithm, which was proposed jointly by the developers of AUGUSTUS and GeneMark-ET.
BRAKER1 has demonstrated a high level of efficiency. The developed program has already been downloaded by more than 1500 different centres and laboratories. Tests of the algorithm have shown that it is considerably more accurate than other similar algorithms. The example running time of BRAKER1 on a single processor is ∼17.5 hours for training and the prediction of genes in a genome with a length of 120 megabases. This is a good result, considering that this time may be significantly reduced by using parallel processors, and this means that in the future, the algorithm might function even faster and generally more efficiently.
Tools such as these solve a variety of problems. Accurately annotating genes in a genome is extremely important – an example of this is the global 1000 Genomes Project, the initial results of which have already been published. Launched in 2008, the project involves researchers from 75 different laboratories and companies. Sequences of rare gene variants and gene substitutions were discovered, some of which can cause disease. When diagnosing genetic diseases, it is very important to know which substitutions in gene sections cause the disease to develop. The project mapped genomes of different people , noting their coding sections, and rare nucleotide substitutions were identified. In the future, this will help doctors to diagnose complex diseases such as heart disease, diabetes, and cancer.
BRAKER1 enables scientists to work effectively with the genomes of new organisms, speeding up the process of annotating genomes and acquiring essential knowledge about life sciences.
Journal information: Bioinformatics
Provided by Moscow Institute of Physics and Technology