Longer DNA fragments reveal rare species diversity

A challenge in metagenomics is that the more commonly used sequencing machines generate data in short lengths, while short-read assemblers may not be able to distinguish among multiple occurrences of the same or similar sequences, making it difficult to identify all the members in a microbial community. In the April 2015 issue of Genome Research, a team including DOE JGI researchers compared two ways of using next generation Illumina sequencing machines to help with this.
Many microbes cannot be cultivated in a laboratory setting, hindering attempts to understand Earth's microbial diversity. Since microbes are heavily involved in, and critically important to environmental processes from nutrient recycling, to carbon processing, to the fertility of topsoils, to the health and growth of plants and forests, accurately characterizing them, as a basis for understanding their activities, is a major goal of the Department of Energy (DOE). One approach has been to study collected DNA extracted from the complex microbial community, or the metagenome, in order to describe its DNA-coded "parts" catalog and understand how microbes respond and adapt to environmental changes. Studying a population rather than an individual raises different obstacles on the path to knowledge. The challenges of assembling genes and genomic fragments into meaningful sequence information for an unknown microbe has been likened to putting together a jigsaw puzzle without knowing what the final picture should look like, or even if you have all the pieces.
"For metagenomics," said Jillian Banfield of the University of California, Berkeley and Lawrence Berkeley National Laboratory's Earth Sciences Division, a longtime collaborator of the DOE Joint Genome Institute (DOE JGI), a DOE Office of Science User Facility, "it is like reconstructing puzzles from a mixture of pieces from many different puzzles—and not knowing what any of them look like." Part of the problem lies in the fact that the more commonly used sequencing machines generate data in short lengths or fragments, on the order of a few hundred base pairs of DNA. Additionally, short-read assemblers may not be able to distinguish among multiple occurrences of the same or similar sequences and will therefore either fail to place them in the correct context, or eliminate them entirely from the final assembly, in the same way that putting together a jigsaw puzzle with many small pieces that look the same, is difficult. The result of this are gaps that indicate not all of the microbes in a community can be identified through the application of environmental genomics.
In a study published on the cover of the April 2015 edition of Genome Research, a team including DOE JGI and Berkeley Lab researchers compared two ways of using the next generation Illumina sequencing machines, one of which—TruSeq Synthetic Long-Reads—produced significantly longer reads than the other. Metagenome data were generated from the Berkeley Lab-led DOE subsurface biogeochemistry field study site in Rifle, Colorado by a Banfield-led team. They evaluated the accuracy of the genomes reconstructed from the sequences produced by the two Illumina technologies to learn more about the microbes present in lower amounts than others and better determine the species richness of the metagenome samples.
The project is part of the Berkeley Lab Genomes-to-Watershed Scientific Focus Area (SFA), which involves over 50 scientists from Berkeley Lab and other institutions including UC Berkeley, Pacific Northwest National Laboratory, Colorado School of Mines, and Oak Ridge National Laboratory. The Genomes-to-Watershed SFA is led by geophysicist Susan Hubbard, the director of Berkeley Lab's Earth Sciences Division. Its goal is to develop an approach for gaining a predictive understanding of complex, biologically based system interactions from the genome to the watershed scale. Jill Banfield is a co-lead of the Metabolic Potential component of this team project, which focuses on characterizing prevalent metabolic pathways in subsurface microbial communities that mediate carbon and electron flux, and using that information to inform genome-enabled watershed reactive transport simulators. Banfield describes the Metabolic Potential component of the SFA effort in a video, and some of her group's other recent groundbreaking subsurface ecogenomic findings associated with this project can be found here.
Revisiting Microbial Communities in Rifle, Colorado
For the study, the team used sediment samples collected from an aquifer adjacent to the Colorado River, which had been used for previous experiments. For one of these earlier efforts the DOE JGI sequenced Rifle Site microbial communities and was able to completely reconstruct a high quality genome of a previously unknown organism from short-read assemblies. Additionally, the findings revealed that many of the bacteria and archaea found in the samples had not been previously recognized or sampled.
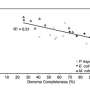
For their study, the researchers compared the sequences and assemblies generated from Illumina's short read technology with the data from the newer, longer-read technology that generates read lengths of up around 8,000 base pairs. They found that the longer reads captured more of the community's diverse species. For instance, using short read technology, they previously identified just over 160 microbial species within a sediment sample. Using the longer-read technology, though, over 400 microbial species from the sample could be phylogenetically classified, though some accounted for just 0.1 percent of the community.
The study's first author, Itai Sharon of UC Berkeley, pointed out that they also identified species that previously failed to assemble due to the presence of closely related species within the sample. These close relatives, accounting for as much as 15 percent of the community, confounded the assembly algorithm. "These populations were pretty much missed by the short read assemblies because assemblers tend to fail at the presence of multiple closely related species and strains. Using algorithms that we developed for analyzing the long reads we were able to reconstruct genome architecture for these populations," he said.
"Extending the analysis further to species with a lower abundance suggests that at least ... 2,100 different species are present," the team reported. "The true number of species is therefore expected to be much higher - probably at the range of several thousands or tens of thousands of different species."
Longer Reads Add Value to Sequencing Capabilities
The difference between the results suggests that the assembly of thousands of rare genomes by short reads failed due to insufficient coverage despite significant sequencing efforts. On the other hand, the longer reads revealed this "long tail" of previously undetected microbial species that were present in very low abundance in the metagenome samples. In addition, short reads assembled poorly for closely related genomes even when enough sequencing coverage is available. Using the long reads it was possible to reconstruct gene order for most of these genomes.
"The availability of both short and long read data allowed us to explore patterns of population diversity, taxonomic diversity, and organism abundance levels using genome sequence information for rare as well as more abundant organisms," the team reported. "Overall, short and long read data provide complementary advantages for metagenome studies, thus making the use of both technologies together more powerful than use of one alone."
DOE JGI Metagenome Program head Susannah Tringe noted that while the Rifle studies came out of a project supported by the Community Science Program (CSP), the longer-read analyses conducted and reported in this study were motivated in part by the DOE JGI's Emerging Technologies Opportunity Program (ETOP). Launched in 2013, the program seeks to develop and support selected new technologies that the DOE JGI could establish to add value to the high-throughput sequencing it currently carries out for its users. "We're not just motivated by wanting to learn about Rifle, but how to use these technologies to learn about microbial communities through ETOP," she added. The inaugural ETOP cycle focuses on six capabilities, one of them a project from Banfield. A key yield from this new sequencing approach is a much more detailed characterization of the microbial communities within a sampled site; not only does this furnish an improved understanding of the processes mediated by microbes taking place at that site—which can include carbon capture, contaminant remediation, or the breakdown of plant and other organic materials which can have bioenergy interest—but also the discovery of new genes and enzymes of interest to DOE missions.
Journal information: Genome Research
Provided by DOE/Joint Genome Institute