SDSC team develops multi-scale simulation software for chemistry research

Researchers at the San Diego Supercomputer Center at the University of California, San Diego, have developed software that greatly expands the types of multi-scale QM/MM (mixed quantum and molecular mechanical) simulations of complex chemical systems that scientists can use to design new drugs, better chemicals, or improved enzymes for biofuels production.
A paper outlining the research, titled 'An Extensible Interface for QM/MM Molecular Dynamics Simulations with AMBER' and conducted by members of the Walker Molecular Dynamics Lab (WMD) at SDSC, was featured on the cover of the January 15th issue of the Journal of Computational Chemistry.
Multi-scale QM/MM computational methods are crucial to advancing the understanding and solution to problems in the chemical sciences, ranging from drug design to renewable energies. This has been recognized with the award of the 2013 Nobel Prize in chemistry for the development of multi-scale models of complex chemical systems.
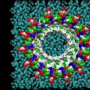
In QM/MM simulations, an accurate but computationally complex and thus time-consuming quantum mechanical model is used to identify important features of the electronic structure of a chemically relevant region. This is required, for example, to describe photo-physical processes or chemical reactions in the active site of enzymes. Effects of the surrounding environment are then included with a computationally less complex classical MM model.
"QM/MM simulations are computationally very demanding compared to purely classical MM simulations," said Ross C. Walker, an SDSC research professor and adjunct associate professor in UC San Diego's Department of Chemistry and Biochemistry. "Access to SDSC's Trestles and Gordon supercomputers and their fast turnaround times were essential to our work. We ran a large amount of jobs to test and validate our implementation at various stages, as well as a large-scale simulation to demonstrate a practical application."
"Our software enables QM/MM simulations with a variety of advanced quantum mechanical models, and by integrating it with the popular AMBER molecular simulation package, which is used by hundreds of academic and industrial research labs, we can reach a very large user base", said lead author Andreas W. Goetz, a research scientist with SDSC and expert in multi-scale modeling. "We're looking forward to many exciting applications that will help scientists in computational chemistry and biophysics understand and predict the behavior of molecular systems at a fundamental level."
Provided by University of California - San Diego