Scientists studying mitochondrial calcium handling yield new disease targets
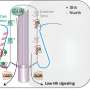
When things go wrong, cells turn to built-in safety mechanisms for survival. One of those mechanisms involves calcium uptake by mitochondria, the energy-producing powerhouses of cells. Long a mystery, new research by scientists at the Temple University School of Medicine (TUSM) Center for Translational Research shows exactly how mitochondria handle damaging excess calcium from the intracellular environment, and how problems with calcium regulation can lead to vascular damage.
"Mitochondrial calcium regulation is essential for cell survival," explained senior investigator Muniswamy Madesh, PhD, Assistant Professor in the Center for Translational Research and the Department of Biochemistry at TUSM. "But the calcium uptake mechanism of mitochondria has been unknown."
In the late 1970s, researchers discovered a mitochondrial calcium influx "set point," a point at which calcium levels become high enough in the cytoplasm (intracellular fluid) to trigger calcium uptake into mitochondria. The set point was determined to be about 3 μM. Dr. Madesh and colleagues previously discovered that below the set point, a protein now known as MICU1 works to suppress calcium influx.
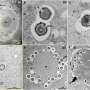
Dr. Madesh's new paper, which appears this week in the journal Cell Reports, is the result of a concentrated effort to identify and describe specific interactions of MICU1. His team began by establishing a novel protein flux dynamics assay, which allowed the researchers to see where MICU1 interactions take place within mitochondria. They then introduced mutations into different regions of the MICU1 protein and investigated how the mutations affected interactions that regulate mitochondrial calcium influx.
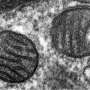
In their protein flux experiments in cells, the team discovered that MICU1 is located in the interior region of the mitochondrion. They also identified the specific regions of MICU1 that determine binding with the uniporter that transports calcium into the mitochondrion.
To characterize the physiological relevance of MICU1, the researchers conducted experiments in mice in which MICU1 was silenced. They found that reduced MICU1 activity resulted in prolonged calcium uptake, chronic oxidative stress, and vascular dysfunction. It also diminished the ability of endothelial cells, which form the inner lining of blood vessels, to migrate, a process necessary for the formation of new blood vessels.
The new work sheds light on ways in which calcium and mitochondrial dysfunction contribute to cell and vascular damage, leading to new opportunities for the discovery of therapies capable of preventing cell injury. According to Madesh, "If we can slow down calcium uptake and protect mitochondria, we may be able to keep mitochondrial energy levels up."
The findings have implications for other research being conducted at Temple's Center for Translational Medicine, where there is particular interest in oxidative damage sustained from conditions such as ischemic reperfusion (when blood flow resumes following a temporary pause, such as during a heart attack).
"Calcium overload and oxidative stress are implicated in cardiovascular and neurodegenerative diseases, aging, and metabolic syndrome," Madesh said. "Calcium overload and oxidative stress is a common feature in disease. It happens all the time."
Journal information: Cell Reports
Provided by Temple University