October 15, 2012 feature
Forensic speciation: Splicing genetic and phylogenic trees of life
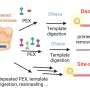
 The eutherian phylogeny estimated using the Bayesian concatenation method implemented in MrBayes. The ML (maximum likelihood) concatenation tree built by RAxML (search algorithm for maximum likelihood) is identical to the Bayesian concatenation tree in topology. Branches of the concatenation tree are coded by the same colors as in the MP-EST tree. The blue asterisks indicate the position of Scandentia (tree shrews), Chiroptera (bats), Perissodactyla (odd-toed ungulates), and Carnivora (carnivores),whose placement differs from the coalescent tree. The Bayesian concatenation tree received a posterior probability support of 1.0 for all nodes. Copyright © PNAS, doi:10.1073/pnas.1211733109")
(Phys.org)—The Tree of Life is a beautiful and elegant metaphor that has proven deceptively difficult to reconstruct. The main culprit may be the overwhelming reliance on so-called concatenation methods, which combine different genes into a single matrix and so force all genes to conform to the same topology. Since these methods do not take into account differences between alternative gene trees, they have been thought to lead to uncertainty or incongruence in the phylogenic tree of the eutherian (placental) mammals. While historically this incongruence had not previously been confirmed by empirical studies, scientists at Shenyang Normal University, Tsinghua University, University of Georgia and Harvard University have recently demonstrated that this is indeed the case – and that concatenation-derived uncertainty may be found in other clades (biological groups derived from a common ancestor) as well. Moreover, the authors suggest that such uncertainty can be resolved by augmenting phylogenomic data with coalescent methods – that is, techniques for dealing with differences in genomic ancestral trees.
The research team – Prof. Shaoyuan Wu, Prof. Sen Song, Asst. Prof. Liang Liu, and Prof. Scott V. Edwards – faced a number of complex issues in conducting their study. "To demonstrate that concatenation methods are actually underlying the controversies in the phylogeny of eutherian mammals, we need to find out what is wrong with concatenation methods," Wu tells Phys.org. "This is a challenging topic since concatenation methods are to date the most dominant approach in the field of phylogenetics." Wu points out that It would be difficult for people to admit that these well-established methods are the cause of controversies in phylogenetic relationships, since for a long time people believe that controversial relationships among eutherian mammals and other clades in the Tree of Life would be resolved as more taxa – groups of one or more populations of organisms – and/or genetic data become available. "However," he notes, "the persistence of these controversies in recent concatenation studies despite the increasing sampling of taxa and genes lead us to believe that something must be wrong with concatenation methods."
Concatenation methods are based on the assumption that all genes have the same or similar phylogenies. However, the team's mammalian data set, gene tree heterogeneity can be found everywhere. While computational simulations have predicted that ignoring gene tree heterogeneity may result in misleading phylogenies, the challenge has been how to empirically test the effect of gene tree heterogeneity on estimating phylogenies.
To address this challenge, Wu explains, the researchers designed their experiment with the innovative approach of using subsampling analysis of loci and taxa – because if gene tree heterogeneity is indeed a confounding factor, the results of the concatenation method are expected to vary according to the histories of the genes represented in a particular subsample. "The subsampling portion of our analysis confirms the prediction that concatenation methods using different subsamples of our data set often conflict with each other, even though metrics such as the bootstrap indicate strong support for each topology – but trees generated from subsamples using the coalescent method are much more topologically consistent."
In addition, he adds, they developed two techniques in this study: estimating the scale of genetic data for accurately resolving a phylogeny based on taxon sampling, and testing if the multispecies coalescent model can explain the observed gene tree data set heterogeneity.
Beyond controversies in eutherian mammal phylogeny, similar phylogenetic controversies also exist in other clades – for example, the relationships among nemerteans, annelids, and molluscs with regards to arthropods. "Because the phylogenic reconstruction in the Tree of Life has so far been mostly based on concatenation methods," Wu adds, "it's likely that concatenation methods are the major cause of phylogenetic incongruence across the Tree of Life." Wu also describes the insights gleaned from the study. Firstly, the researchers showed using coalescent methods to deal explicitly with gene tree heterogeneity is preferable to applying concatenation methods to data sets with high gene tree heterogeneity. A second insight was that it is also critical to gather a sufficient number of loci to obtain an accurate phylogeny for mammals and other clades despite the importance of taxon sampling for phylogenetic analysis. "For example," Wu illustrates, "the intensive taxon sampling employed in recent research1 cannot compensate for the effect of insufficient genetic sampling in their data set."
Finally, Wu notes, incomplete linage sorting (ILS), a major source of gene tree heterogeneity, is relevant to deep-level phylogenies. "This is in contrast to the conventional assumption that ILS is only relevant to recent radiations," he stresses. "ILS is prevalent in coding sequences, which is in contrast to recent suggestion that coding sequences may be less subject to ILS than noncoding sequences due to frequent selective sweeps, which tend to remove ILS."
Wu expands on the paper's key conclusion – namely, that such incongruence can be resolved using phylogenomic data and coalescent methods that deal explicitly with gene tree heterogeneity. "The prevalence of gene tree heterogeneity in genomic data indicates that a good phylogenetic method should take this complexity into account when inferring species phylogenies," he points out. "It's clear that concatenation methods, which assume gene tree homogeneity, do not fit the complexity of phylogenetic reality – that is, that gene tree heterogeneity is common among all genes and taxa. In contrast, the multispecies coalescent model can explain 77% of gene tree heterogeneity observed in the mammal data set, indicating that the coalescent approach indeed gives a better picture of complex phylogenetic reality when gene tree heterogeneity is prevalent in the data sets."
Delving deeper, Wu notes that the erratic behavior of concatenation methods confirms that concatenation methods are not suitable for genomic data, which possess substantial levels of gene tree heterogeneity. "The robustness of coalescent methods to variable gene and taxon sampling demonstrates that coalescent methods are superior to concatenation methods in building species phylogenies based on phylogenomic data by accommodating gene tree heterogeneity – and the data suggests controversial relationships in the Tree of Life can be resolved as more data are collected. In other words, resolving the phylogeny of eutherian mammals and other clades in the Tree of Life will require a large amount of data at genomic scale."
To extend the current study, the scientists' next research step is to assess the suitability of tree-building models for different types of genomic data, and to examine how different characteristics of genomic data would affect the performance of tree-building methods. Moreover, the paper has implications for other areas of research as well. "Besides the field of evolutionary biology," Wu concludes, "a well-resolved phylogeny has important applications in the studies of comparative genomics and biomedical sciences. The major contribution of this study is to provide an example and a roadmap to help researchers to build accurate phylogenies using genomic data, which will certainly benefit studies in these areas."
More information: Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model, PNAS September 11, 2012, vol. 109 no. 37 14942-14947, doi:10.1073/pnas.1211733109
1Related: Impacts of the Cretaceous Terrestrial Revolution and KPg Extinction on Mammal Diversification, Science 28 October 2011: Vol. 334 no. 6055 pp. 521-524, doi:10.1126/science.1211028
Journal information: Science , Proceedings of the National Academy of Sciences
Copyright 2012 Phys.org
All rights reserved. This material may not be published, broadcast, rewritten or redistributed in whole or part without the express written permission of PhysOrg.com.