September 24, 2012 feature
Making a molecular micromap: Imaging the yeast 26S proteasome at near-atomic resolution
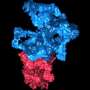
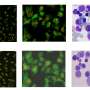
 single particle reconstruction of the Saccharomyces cerevisiae 26S proteasome without imposed symmetry. The density is displayed as an isosurface from two different views, on the right colored according to the local resolution as indicated by the color key. Five different slices from the upper RP (A–E) are shown and compared to the counterparts in the lower RP (A’–E’) by difference images. Copyright © PNAS, doi:10.1073/pnas.1213333109")
(Phys.org)—Biological systems are characterized by a form of molecular recycling – and proteins do not escape this fate. In particular, unneeded or damaged proteins biochemically marked for destruction undergo controlled degradation by having their peptide bonds broken by proteasomes. Recently, scientists at the Max-Planck Institute of Biochemistry in Germany used cryo-electron microscopy (cryo-EM) single particle analysis and molecular dynamics techniques to map the Saccharomyces cerevisiae 26S proteasome. (Cryo-EM is a form of transmission electron microscopy where the sample is studied at cryogenic temperatures, which unlike X-ray crystallography allows researchers to observe specimens in their native environment without the need for staining or fixing. S. cerevisiae is the yeast species commonly known as baker's or brewer's yeast.) The researchers then used this map to build a near-atomic resolution structural model of the proteasome. The Max Planck team showed that cryo-electron microscopy allowed them to successfully model the 26S core complex where X-ray crystallography studies conducted over the past 20 years have not.
Dr. Friedrich Förster relates the main challenges the researchers faced to Phys.org, starting with achieving automated single particle acquisition. "The aim of automated single particle acquisition is to collect large amounts of high-quality electron micrographs," Förster begins. "The microscope settings need to be kept essentially invariable over a period of several days, which requires constant adjustment of imaging parameters." These key parameters, he explains, are image defocus (electron micrographs are not acquired exactly in focus, but with a chosen defocus of typically 1-3 micrometers), eucentric height (an electron microscopy grid is fairly "bumpy" on a micrometer scale, which needs to be compensated for by vertically adjusting the height of the specimen), and illumination settings (lens currents vary to some extent over time, yielding illumination variations that need to be corrected for). "Moreover, "Förster adds, "it needs to be ensured that the whole EM grid is covered with non-redundant images – which is challenging due to specimen movements – and acquisition of 'garbage' needs to be minimized (for example, an EM grid typically contains large areas that are intransparent to the electron beam)."
The second challenge, Förster continues, was acquiring the cryo-EM structure of the S. cerevisiae 26S proteasome. "The particular challenge of the 26S proteasome is that it cannot be purified to very high concentrations and homogeneity as it often disassembles into smaller building blocks. Hence, an electron micrograph contains relatively few individual particles and many micrographs need to be acquired." The next step – using the proteasome map building a near-atomic resolution model – has a different issue. "One challenge is the sheer size of the complex: Overall, 33 different subunits are fitted into the map." This is a number exceeding almost all other structures studied by cryo-EM single particle analysis with the notable exception of the ribosome. "A further problem is that all models are refined starting from homology models, as opposed to X-ray crystallographic structures of the same proteins in different conformations. Homology models, in particular when the sequence similarity is 30% or lower, typically deviate substantially from the true protein structures. Inaccuracies tend to be particularly large for side-chains. Due to these substantial inaccuracies of the initial models the refinement of the models is cumbersome and requires high scrutiny."
Another issue is using the map to assign α-helices throughout the entire map, "The assignment of α-helices results from the molecular dynamics-based flexible fitting," Förster notes. "We estimate our model to be very reliable for α-helical segments, which are well-resolved in the EM map, whereas accuracy is lower for other parts of the models. Luckily, the 26S proteasome consists mostly of helices."
Yet another issue was determining the architecture of the Rpn8/Rpn11 heterodimer, which was entirely unknown prior to their study. "To determine its architecture it was essential to reconstruct the 26S without symmetry, since this symmetry does not seem to apply to high resolution," Förster points out. "This resulted in a reconstruction with sufficient resolution to localize secondary structure elements in this specific area of the 26S holocomplex."
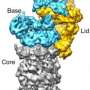
Interestingly, Förster adds that in this work they did not develop new methodology. "Rather, we applied existing methodology from different areas – data acquisition, single particle reconstruction, and computational modeling – and combined them in an efficient pipeline. Probably, the key insights that we got from this work is the structural organization of the proteasome lid – namely, that it's held together by a large helical bundle – and how the deubiquitylating site of Rpn11 is positioned at the mouth of the AAA-ATPase." Deubiquitylating enzymes (DUBs) can hydrolyze a peptide, amide, ester or thiolester bond at the C-terminus of ubiquitin (UBIQ), including the post-translationally formed branched peptide bonds in mono- or multi-ubiquitylated conjugates.
Moving forward, Förster tells Phys.org, there are a number of innovations they would like to apply to the current experimental design. "It would be desirable to improve the resolution of the 26S cryo EM map further to improve the accuracy of our model," he illustrates. "Key to this improvement will be to tackle two things: the structural heterogeneity of particles –different structural modes underlie the observations – and removal of non-informative particles from datasets, since for not entirely understood reasons many particles make reconstructions worse rather than better)." For further data analysis they plan to integrate more elaborate classification methods to address these issues.
"After having obtained a first draft of the 26S proteasome structure," Förster continues, "the next big challenge is to understand how it works. In-depth analysis of the different conformational states that are present in our dataset will provide some clues to the conformation space. Cryo-EM studies of the 26S proteasome engaged with substrates and different biochemical and chemical treatments will then provide more specific insights into the structural reorganizations of the 26S proteasome during its functional cycle."
Förster also mentions the other areas of research that might benefit from their findings. "Firstly, our results will be of great interest to scientists working on the ubiquitin-proteasome pathway, the most important route for regulated protein degradation in all eukaryotic cells. For example," he illustrates, "the model will be invaluable to interpret data on the assembly process of the 26S proteasome and to study interactions of the 26S proteasomes with other molecules." Moreover, he points out, the 26S proteasome important as a drug target: The proteasome inhibitor Velcade is a successful anti-cancer drug, and the proteasome is also a promising target of drugs against neurodegenerative diseases1. "Therefore, the 26S proteasome structure," Förster concludes, "will also be of interest to pharmaceutical and medical scientists."
More information: Near-atomic resolution structural model of the yeast 26S proteasome, PNAS September 11, 2012 vol. 109 no. 37 14870-14875, doi:10.1073/pnas.1213333109
1Related: Enhancement of proteasome activity by a small-molecule inhibitor of USP14, Nature 467, 179–184 (09 September 2010), doi:10.1038/nature09299
Journal information: Proceedings of the National Academy of Sciences , Nature
Copyright 2012 Phys.org
All rights reserved. This material may not be published, broadcast, rewritten or redistributed in whole or part without the express written permission of PhysOrg.com.