The controlled cell
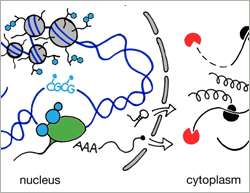
(Phys.org)—An interdisciplinary effort at the Friedrich Miescher Institute for Biomedical Research (FMI) addressed the question of how mRNA content, which is translated into proteins, is regulated in the cell. Supported by the Swiss initiative for systems biology (SystemsX.ch), experimental biologists and computer scientists teamed up to contrast epigenomic data of histone modifications with post-transcriptional read-outs. Their study, published as a featured article in the journal Molecular Systems Biology, gives answer to a quantitative question in gene regulation.
Traditionally gene regulation was described as a process driven by the action of transcription factors (TF) turning genes "on" or "off". In more recent years chromatin has been described as an important structure controlling accessibility of relevant TF binding sites. What is more, open or closed chromatin states were linked to specific modifications on histones. The advent of chromatin biology brought thus a new layer of control to the traditional view of gene regulation. Finally, with the discovery miRNAs as an extensive class of regulatory RNAs in 2001, a whole new field of research challenged the exclusive role of transcriptional control in gene regulatory processes. Both chromatin and RNA biology provide thousands of examples linking development and disease phenotypes to shifts in RNA synthesis or decay, respectively. But the question remains: "Which process is the key driver?"
The abundance of each mRNA depends on both, its synthesis and its decay. Today, we have a good understanding of how RNA transcription is regulated and we know many factors that contribute to the decay of a transcript. What we do not know is what pathway is the main contributor to steady state mRNA levels. A recent publication by the group of Dirk Schübeler, group leader at the Friedrich Miescher Institute for Biomedical Research, addresses this question on a genome wide scale and provides a clear answer: the main driver is transcription.
Schübeler's laboratory works with a cell culture system, differentiating mouse embryonic stem cells (ESCs) to neurons. This allows monitoring two conceptually different cell stages: dividing and post-mitotic cells. In both cell types the scientists established high resolution genome wide maps of histone modifications using chromatin immunoprecipitation followed by deep sequencing (ChIP-seq). Additionally, they sequenced the transcriptomes and correlated the abundance of each mRNA to histone modifications at the corresponding locus in the DNA. Some of these histone modifications have been related to an open, some to a closed chromatin state suggesting a relationship between chromatin state and transcription. Using a simple modeling approach, the scientists could indeed predict up to 84% of the variance in mRNA levels.
"We were surprised to see that chromatin marks alone can approximate RNA levels so well, especially given the measurement error inherent to each experimental readout," said Sylvia Tippmann, first author of the study and PhD student supervised by Schübeler and Michael Stadler, who heads up Computational Biology at the FMI.
The error of a measurement is indeed important: technical noise is introduced by the variability in the experiments and, moreover, a biological system never behaves exactly the same from one measurement to the other. The higher the reproducibility between repeated experiments, the smaller is the error. In collaboration with Erik von Nimwegen's bioinformatics group from the Biozentrum of the University of Basel, the scientists were able to calculate an upper bound of how closely they can approximate mRNA levels based on repeat measurements. In other words, they calculated what percentage of the mRNA level is obscured by the measurement error.
Given the difference between the achieved correlation with RNA levels by chromatin measurements and this upper bound, only little variance in mRNA levels remained unexplained. The researchers went on to test whether this remaining variance can be explained by mRNA half-life and more specifically by miRNA induced mRNA decay. Although minor, an effect of half-life on the abundance of mRNA was measurable. However the influence of miRNAs on their potential target genes could only be quantified for a selected group of very likely miRNA target genes.
"It is important to keep in mind that our study is a quantitative account, not an assessment of importance of biological processes. There are genes that are mainly regulated on the level of decay. However, on average, steady-state mRNA levels are largely determined by transcriptional regulation," emphasizes Tippmann.
More information: Tippmann SC, Ivanek R, Gaidatzis D, Schöler A, Hoerner L, van Nimwegen E, Stadler PF, Stadler MB, Schübeler D (2012) Chromatin measurements reveal contributions of synthesis and decay to steady-state mRNA levels. Mol Syst Biol 8:593. www.ncbi.nlm.nih.gov/pubmed/22806141
Journal information: Molecular Systems Biology