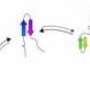
Scientists study genomes to reveal contacts, refine methods for protein-folding prediction
 and estimate amino acid contacts. The contacts, shown in red, orange and yellow in the 3-D structure, serve as key elements to fold the protein into its native state. (Credit: Onuchic Research Group)")
José Onuchic has become an expert at connecting the dots, but finding connections merely implied by the dots … well, that's quite a trick.
The Rice University biophysicist and his team have created a tool to do just that for proteins and, in the process, have advanced the art of predicting their form and function.
In this case, the dots are amino acid molecules known as residues that link together in chains to form proteins. Proteins are the workhorses that carry out the biological tasks essential to every living thing, but before they can go to work, they fold. Each protein has its own characteristic, folded shape, and various diseases, including cancer, have been linked to proteins that misfold or otherwise misbehave.
As computers grew more powerful over the past three decades, scientists have created many methods to predict how a particular chain of residues is likely to fold and the purpose the resulting protein serves.
Onuchic and colleagues at the Center for Theoretical Biological Physics have developed a tool, known as direct coupling analysis-fold (DCA-fold), that enhances existing methods. Details of their research appear today in the online version of the Proceedings of the National Academy of Sciences (PNAS). The center is currently based at the University of California at San Diego (UCSD) but is relocating to Rice's BioScience Research Collaborative.
While most protein-folding researchers look at the sequence of amino acids in a protein, either through X-ray crystallography of folded proteins or through computer simulations, Onuchic and his colleagues stepped back to look at the DNA sequences that serve as the blueprints for the proteins. By exploiting the increasingly large database of genomic sequence information, they're able to increase the accuracy of predicting the structures of folded proteins.
They start by finding points in the protein-encoding genome sequences that appear to change at the same time, even though they may be separated by great distances along the chain. The implication, Onuchic said, is that at some point in the protein's evolutionary history, the amino acids made contact, liked what they saw and kept in touch. In more technical terms, a benefit to the protein's purpose was realized and conserved.
"A lot of the decisions made through the biological process don't depend on a strong partner," said Onuchic, Rice's Harry C. and Olga K. Wiess Chair of Physics and a professor of physics and astronomy, chemistry, biochemistry and cell biology. "It's like the protein comes, kisses you once, and goes away; it's what we call very weak interaction, which we're never going to be able to see with current methods.
"But those weak interactions can cause a conformational change, transfer a phosphorus or start an entire cascade of signals," he said.
Onuchic, lead authors Joanna Sulkowska and Faruck Morcos, postdoctoral researchers at UCSD, and their colleagues looked deeply into the genomes of bacteria to pull 15 protein models; the scientists took from them about 1,000 distinct protein-coding sequences, enough for DCA-fold to be statistically accurate.
"When you look at the evolution of a particular protein in these bacteria and see just one residue change from one sequence to the next, that's probably random," Onuchic said. "But if two change at the same time, the probability is that they're changing together. It's a good sign that these things probably interact with each other."
Spotting those interactions is difficult in the proteins themselves, at least with current methods, he said. Crystallization, for instance, freezes a protein in time but provides no evidence of interactions that happened on the way to the finished product. And while computer simulations that align protein sequences are improving, their accuracy is not as good as it needs to be, he said.
But direct coupling analysis of protein-coding genes spots positions in sequences conserved across genomes that change together – a change that could only happen through mechanical contact.
DCA-fold finds those subtle interactions that other methods miss. Energy landscape theories developed by Onuchic and his team predict how those interactions nudge the protein through its process. The combined result eliminates possibilities from the range of forms a protein might take.
Onuchic sees that as a way to pull a signal from all the noise.
"The entire game here is to show that by adding the genomic data to folding simulations, we can aid in structure prediction," Onuchic said. "Here, we get at least 1,000 bacterial sequences that are part of the same protein family but are not necessarily structurally similar. Then we compare the sequences and figure out which pairs of amino acids change at the same time. Although previous correlation methods could give approximate answers, our model is much more accurate.
"Once we know there's a high probability that these two amino acids came together at some point, we're constrained. If the sequence data tells me we can have structure a, b or c, we can then look to see which is consistent with the pair of contacts we now know about from the genomic data and eliminate the wrong predictions."
The new paper was one of three published by Onuchic and his colleagues this month. A second, also in PNAS, examines proteins that not only knot but sometimes form slipknots. (Read about that here.) The third, which appeared in PLoS One and on which Onuchic was a co-author, analyzes how the Interleukin-1 beta protein resolves the conflicting energeticdemands of its residues as it folds.
All of this work brings Onuchic and his colleagues from UCSD who are joining Rice closer to the purpose of their move: To bring all the data they've accumulated on biological systems to bear on the treatment of cancer.
Eukaryotic cells like those in humans (and unlike those in simple bacteria) produce many thousands of proteins; with rapid advances in the capability to sequence genomes, the researchers hope to apply their technique to look for what causes diseases and how to cure them.
"I think DCA has enormous potential," Onuchic said. "The amount of genome data that's being created is growing exponentially. In the future, if we're patient, instead of comparing sequences of different bacteria as we do here, perhaps we can do time evolution of sequences. Then we can figure out if a particular disease mechanism comes from a pair of residues that switch at the same time – and instead of looking at different animals, we can see changes in one patient over time."
Co-authors of the paper are Terence Hwa, a professor in the Department of Physics at UCSD, and Martin Weigt, a professor at Pierre and Marie Curie University, Paris.
More information: Read the open-access paper at www.pnas.org/content/early/201 … 864109.full.pdf+html
Journal information: Proceedings of the National Academy of Sciences
Provided by Rice University