Rewriting the Organofluorine Playbook
2, in the gas-phase by low-temperature photoelectron spectroscopy and in solution by cyclic voltammetry. Image by Alexey A. Popov,")
(Phys.org) -- Sometimes it is easy to overgeneralize, to conclude that simply because a group of things are pretty much all the same, they're identical in all respects, even interchangeable. But such assumptions can cause a lot of problems, not just in everyday life but also in science. One example is the findings of a research team whose experiments at two U.S. Department of Energy Office (DOE) of Science facilities, including the Advanced Photon Source (APS) have shaken up the field of organofluorine chemistry by challenging some long-held theoretical assumptions, and because of their implications for the practical application of materials based on certain important molecules. Their published study was the cover article May 2012 issue of the journal Chemical Science.
The highly complex and specialized realm of organofluorine chemistry involves the study and application of organic compounds based on the carbon-fluorine bond. Organofluorines are used in a dizzying array of applications, including solvents and nonstick coatings, blood substitutes, anesthetics and various other drugs, lubricants, and even organic photovoltaic (OPV) devices such as highly efficient solar cells.
Because the electronic properties of a particular compound are crucial to understanding and controlling its application, chemists have devised some handy mathematical shortcuts for determining and comparing the properties of related organofluorines.
Basically, the idea is that by experimentally determining the value of one of these properties, the others could be derived simply by using a set of simple linear equations.
But the team of researchers in this study demonstrated that it's not that simple.
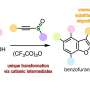
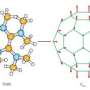
After preparing a series of seven 1,7-C60(RF)2 organofluorine compounds and carefully studying the electron affinities, first-order reduction potentials (E1/2(0/-), and DFT-predicted E(LUMO) values of each, the team, from Colorado State University, The University of Chicago, the Dresden University of Technology, Washington State University, the Liebniz Institute for Solid State and Materials Research, Argonne National Laboratory, and Pacific Northwest National Laboratory found that the convenient and generally assumed correlations among these properties were not present.
The research team, led by some expert organofluorine chemists, conducted advanced x-ray crystallography at ChemMatCARS x-ray beamline 15-ID-B.
The seven compounds studied (RF = CF3, C2F5, n-C3F7, i-C3F7, n-C4F9, s-C4F9, and n-C8F17) were chosen because of their potential use as electron acceptors in organic photovoltaic materials.
“In previous work we had studied how different numbers of a single type of perfluoroalkyl [PFA] group, CF3 [trifluoromethyl[, in different relative positions on the [fullerene] cage will change the electron accepting properties,” said co-author and team member Steven H. Strauss of Colorado State University.
“In this paper, we wanted to know whether, if we have the same structure and the same relative positions, what happens if we change the PFA group from a short group like trifluoromethyl CF3 to a long group like n-C8F17. Is there a difference in the electron withdrawing properties?”
The common wisdom that has guided the design and development of most organofluorines is that such similar compounds will also share more or less the same electrochemical properties. For example, determining the first reduction potential (E1/2(0/-) of a particular molecule will also allow its electron affinity to be estimated.
“But here's an example of a series of compounds where the correlations don’t work, where the solution electrochemistry would lead one to believe that these compounds are virtually the same, and yet they're not,” Strauss said.
At first, the seven PFA compounds displayed virtually identical first reduction potentials at the ±10-mV level of uncertainty.
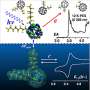
The researchers confirmed the molecular structures and PFA positions with low-temperature anion photoelectron spectroscopy carried out at the Environmental Molecular Sciences Laboratory (EMSL) at the DOE’s Pacific Northwest National Laboratory, and very precise x-ray crystallographic studies at the ChemMatCARS facility at the APS.
“The collaboration with Yu-Sheng Chen at ChemMatCARS was important for the project,” said team member and co-author Olga V. Boltalina. “The facilities he developed can handle the smallest crystals of fullerenes and his expertise in x-ray characterization and solid-state packing of molecules were invaluable.”
But when the experimenters measured electron affinities of the compounds in the gas phase, they found them to be quite different. The researchers were also surprised by a significant lack of correlation between the measured E1/2(0/-) and E(LUMO) values predicted with DFT calculations.
The team's findings have shaken up the field of organofluorine chemistry not only because they have overturned some long-held theoretical assumptions, but also because of their implications for the practical application of materials based on such molecules, such as OPVs.
“This was a real surprise to us and, I think, to the community,” Strauss said. “It points out that it's going to be risky to take results from experiments in one phase (e.g., solution, gas phase) and draw conclusions about the molecules in another phase (e.g., solid thin films).”
The work demonstrates that greater precision and accuracy in measurement are going to be necessary to create fullerene materials with the desired electrochemical properties.
“This is basically an alert to the community that one really has to be cautious that when trying to design a system, one is going to have to measure a number of different electrochemical properties or electron acceptor properties to get a real idea of how a particular compound might result in a particular efficiency in a solar cell,” Strauss said.
The next step, said Boltalina, will likely involve studying the solid state electronic signature of these materials: “That way, we'll get into all three phases.”
"When we get solid state measurements, we can now correlate them with the gas phase and solution phase,” Strauss said.
Armed with that information, Strauss and his team will be able to “collaborate with those that are actually making devices, provide them with our compounds, and see how well they work or don't work. Either way we learn something about what's important in making better devices.”
More information: Igor V. Kuvychko1, James B. Whitaker1, Bryon W. Larson1, Travis C. Folsom1, Natalia B. Shustova1, Stanislav M. Avdoshenko, Yu-Sheng Chen, Hui Wen4,5, Xue-Bin Wang, Lothar Dunsch6, Alexey A. Popov, Olga V. Boltalina, and Steven H. Strauss1*, “Substituent effects in a series of 1,7-C60(RF)2 compounds (RF = CF3, C2F5, n-C3F7, i-C3F7, n-C4F9, s-C4F9, n-C8F17): electron affinities, reduction potentials and E(LUMO) values are not always correlated,” Chem. Sci. 3, 1399 (2012). DOI:10.1039/c2sc01133f
See also: “Calling familiar assumptions into question results in better materials design.”
Journal information: Chemical Science
Provided by Argonne National Laboratory