Researchers create first custom designed protein crystal

Protein design is technique that is increasingly valuable to a variety of fields, from biochemistry to therapeutics to materials engineering. University of Pennsylvania chemists have taken this kind of design a step further; using computational methods, they have created the first custom-designed protein crystal.
Picking an ambitious design target with challenging features, the researchers' success bodes well for the technique's use in better understanding proteins' makeup or using their self-assembling properties in making new materials with unique properties.
The research was conducted by professor Jeffrey G. Saven, postdoctoral fellow Christopher J. Lanci and graduate student Christopher M. MacDermaid, all of the Department of Chemistry in Penn's School of Arts and Sciences. Also contributing to the work were Seung-gu Kang and Xi Yang, formerly of the chemistry department, and Rudresh Acharya, Benjamin North, X. Jade Qiu and William F. DeGrado, formerly of Penn's Perelman School of Medicine's Department of Biochemistry and Biophysics.
The team's research was published in the journal Proceedings of the National Academy of Sciences.
Proteins are folded strings of molecular building blocks known as amino acids; their different functions are determined by their sequences of amino acids and the shapes they take when folded. As proteins are involved in most biological processes, determining sequences and structures is crucial to many scientific undertakings, such as understanding disease mechanisms or designing drugs to disrupt them.
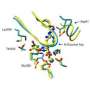
To determine protein structures, scientists use crystals, which consist of many copies of a single protein lined up and stacked together. By irradiating the crystal with powerful X-rays, they can measure the way the light diffracts off the atoms and piece together the protein's overall three-dimensional shape and composition. Most proteins don't naturally crystalize, however, and making crystals of sufficient quality to do diffraction studies is a hit-or-miss process that can take years of painstaking work.
Protein crystals are also attractive as a nano-scale building material, as their properties, particularly their exterior surfaces, are highly customizable. However, bioengineers run into the same hurdles as crystallographers; making a protein crystal with a particular structure is a complex, hard-to-predict task.
"People have designed crystals out of smaller, much less complex molecules than proteins, but protein design is much more subtle," Saven said. "It's a complicated symphony of intermolecular interactions."
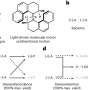
As accounting for these many interactions is one of the principal challenges behind designing a protein crystal, the researchers selected a complicated, honeycomb-shaped target to show their process could be widely applied.
That process involved finding the right protein and designing how copies of it will interact with each other. To tackle the tremendous number of variables involved, the researchers developed a theoretical method and computer algorithm to search through potential proteins for ones that could crystalize into their target.
The researchers targeted a crystal built using a relatively small protein containing a sequence of 26 amino acid positions. The researchers assigned specific amino acids to eight of the positions, but, with 18 different types of amino acid to choose from for each of the remaining 18 slots, the algorithm addressed well more than 1022 potential combinations. On top of that, the researchers had to account for other characteristics, such as the spacing between proteins and their orientation with respect to one another. These variables multiplied the already astronomically large number of possibilities, so maximizing the efficiency of the search was a priority.
"We worked on synthesizing both of those steps, doing the characterization of structure and the sequence at the same time," Saven said. "As we move through this process, we eliminate things that will never work, such as proteins where atoms overlap in space or where amino acids don't fit into a given site. At the same time, we identify proteins that, as you vary the structure, are likely to yield a crystal."
"Combining theory with recent advances in computer hardware have really allowed us to consider thousands of candidates, instead of a just a handful," MacDermaid said
After a full day of computation, the researchers' algorithm produced a handful of promising candidates. Critically, these proteins had very different sequences from one another. Even with their computational approach, finding the protein that would crystalize into their target involved trial and error.
"One reason to consider a very broad range of candidate proteins is that in nature you have proteins that are the same across organisms — the same name, structure and function — but their sequences are often very different," Saven said. "Nature finds many different possible solutions to the same problem, so we wanted to develop methods that allow us to say something about many disparate possible solutions."
This approach is particularly important when considering the eventual applications of a protein crystal. One sequence may crystalize perfectly but be toxic to cells, making it difficult to produce or unusable for a biotechnological application. A non-toxic sequence may form the crystal but only in tiny quantities.
"It's an important part of our algorithm that you don't end up with single sequence and put all of your eggs in one basket," Lanci said. "You get a large landscape of possibilities, improving the odds that you'll find one that can overcome all the experimental challenges you can't control for."
Beyond presenting multiple candidates, the computational approach has the advantage of finding proteins that will produce diffraction-quality crystals. The researchers saw their candidate proteins begin to crystalize within hours, a process that can often take weeks or months.
Though they achieved their goal, the target crystal the researchers produced is just a proof of concept. The researchers' algorithm represents an important tool kit for better understanding the principles behind protein crystallization and the many variables involved. By adding that understanding back into the algorithm, researchers will be able to search for candidate proteins more efficiently.
"There's still much we don't know about the interactions that govern crystallization," Saven said. "With this technique, we can explore what those interactions are or how we might take an existing protein and engineer those interactions so we get much better structures."
Provided by University of Pennsylvania