Infrared spectroscopy allows scientists to analyze protein structure on ultrafast timescale

Proteins can take many different shapes, and those shapes help determine each protein’s function. Analyzing those structures can tell scientists a great deal about how a protein behaves, but many of the methods now used to study structure require proteins to be crystallized or otherwise altered from their natural state.
Now, MIT researchers have developed a way to analyze proteins that doesn’t require any pre-treatment. The technique is also extremely fast, allowing scientists to see, for the first time, how a protein changes its shape over picoseconds, or trillionths of a second.
The researchers, led by chemistry professor Andrei Tokmakoff and postdoc Carlos Baiz, describe their new technique this month in the journal Analyst. Their approach builds on a technology known as two-dimensional infrared spectroscopy, which works by shining pulses of infrared light on a molecule and measuring the resulting molecular vibrations. In the new paper, the researchers came up with a way to analyze that data and correlate it with common structural elements found in proteins.
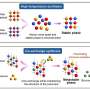
Once assembled, proteins tend to fold into one of two secondary structures, known as alpha helices and beta pleated sheets. In this study, the researchers distinguished between those two structures by examining how bonds between carbon and oxygen — found in each of the amino acids that make up proteins — vibrate when exposed to infrared light.
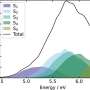
In an alpha helix, the carbon-oxygen bonds run parallel to the protein’s backbone; in a beta sheet, those bonds are perpendicular to the sheet. Because of that difference, the bonds vibrate at different frequencies when struck with infrared light. This allows the researchers to calculate the percentage of the amino acids that belong to a helical structure and the percentage that form a beta sheet.
The researchers confirmed the accuracy of their calculations by analyzing a set of proteins whose structures are already known. Their method does not currently reveal the exact structure of a protein, but the researchers are working on ways to determine the arrangements of the sheets and helices from the spectroscopic data.
“In principle, the full structure of the protein is represented in the spectrum. The trick is how to get out the information,” says Baiz, lead author of the paper.
One way to do that is to analyze data from a broader range of infrared wavelengths. The researchers are also developing methods to get information about other bonds within the amino acids.
Because the new method can be performed over millionths of a second, it can be used to study how proteins fold and unfold when denatured by heat. After hitting a protein with a laser blast to heat it up, the researchers can capture a series of snapshots of how the protein unfolds over this very short time period.
“This is the first method that will allow us to take snapshots of the structure of the protein as it’s denatured,” Baiz says. “Usually the way people look at proteins is they start with the unfolded state and they end up with the folded state, so you have two static structures. What we can do now is look at all the structures along the pathway.”
Munira Khalil, an assistant professor of chemistry at the University of Washington, says the ability to track structural changes over time is the technique’s biggest strength. “One big question is how do proteins fold — at what point does it go from a completely disordered structure to an ordered structure?” says Khalil, who was not involved in this research.
This would be particularly useful for studying proteins that cause disease when misfolded, such as the tau protein found in patients with Alzheimer’s disease and the prion that causes Creutzfeldt-Jakob disease.
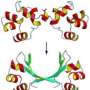
The method can also measure the structural changes that occur as proteins bind to each other. “If the protein is like a rock, and doesn’t change, then it’s never really going to bind its target or do anything. Those are the types of processes we can look at — the conformational changes that drive biological function,” Baiz says.
Provided by Massachusetts Institute of Technology
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.