Study reveals how lung cancers evolve in response to targeted treatment
A detailed analysis of lung tumors that became resistant to targeted therapy drugs has revealed two previously unreported resistance mechanisms. In a report in the March 23 Science Translational Medicine, investigators from the Massachusetts General Hospital (MGH) Cancer Center also describe how the cellular nature of some tumors actually changes in response to treatment and find that resistance-conferring mutations can disappear after treatment is discontinued. The findings support the importance of monitoring the molecular status of tumors throughout the treatment process.
"It is really remarkable how much we oncologists assume about a tumor based on a single biopsy taken at one time, usually the time of diagnosis," says Lecia Sequist, MD, of the MGH Cancer Center, lead author of the report. "Many cancers can evolve in response to exposure to different therapies over time, and we may be blind to the implications of these changes simply because we haven't been looking for them."
Non-small-cell lung cancer (NSCLC) is the leading cause of cancer death worldwide, and in about 12 percent of patients the tumor is driven by a mutation in the epidermal growth factor receptor (EGFR), which stimulates uncontrolled cellular growth. A group of targeted drugs called tyrosine kinase inhibitors (TKIs) block EGFR activity and can halt the growth of tumors driven by such mutations. But in most patients with cancers that respond to TKIs – the best known of which are erlotinib (Tarceva) and gefitinib (Iressa) – resistance develops after about a year of treatment and tumors resume growing.
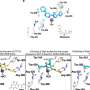
Two mechanisms for this resistance have been identified – a second EGFR mutation that blocks TKI activity and overproduction of the MET oncogene. There also have been reports of resistant tumors regaining sensitivity to TKIs after a drug-free interval. To better understand the molecular basis for TKI resistance, the research team did a comprehensive analysis of both the genotype and the phenotype or physical characteristics of tumor samples from 37 NSCLC patients, samples taken both before TKI treatment was initiated and when resistance first appeared. The results validated the previously reported mechanisms and identified two more genetic changes – mutations in another oncogene called PIK3CA and overproduction of the EGFR molecule itself.
In samples from five patients, the tumors actually transformed into small-cell lung cancers (SCLC), which can respond to other, more traditional chemotherapy drugs. In two patients the appearance of tumor cells changed from that of the epithelial cells that line bodily surfaces and cavities to that of mesenchymal or connective tissue. A few isolated instances of those changes have been reported previously, and their appearance in this study supports a role as resistance-conferring alterations, the authors note. Over a two-year period repeat biopsy samples were taken from three patients whose tumors developed resistance to TKI treatment during that time. Those samples showed that both genetic and phenotypic resistance mechanisms disappeared when treatment was discontinued, providing a mechanism for the previously reported re-sensitization to TKI therapy.
"Our findings suggest that, when feasible, oncogene-driven cancers should be interrogated with repeat biopsies thoughout the course of the disease," says Sequist. "Doing so could both contribute to greater understanding of acquired resistance and give caregivers better information about whether resumption of targeted therapy or initiation of a standard therapy would be most appropriate for an individual patient."
Adds senior author Jeffrey Engelman, MD, PhD, of the MGH Cancer Center, "Now we need to better understand the molecular changes that underlie the transitions from NSCLC to SCLC and from epithelial to mesenchymal morphology. We also need to further evaluate those cancers - eight in this group - for which no resistance mechanism has been identified." Both Engelman and Sequist are assistant professors of Medicine at Harvard Medical School.