Developing a library of cancer proteins

Ten years after the first human genome was sequenced, science is about to reach a new milestone. Researchers are now turning their attention to the products which use genes as instructions for their assembly: the proteins. There are 21,000 genes in the entire human genome, and the scientific research team around Matthias Mann from the Max Planck Institute for Biochemistry in Martinsried estimates that approx. 12,000 proteins are produced in typical human cells. This implies an enormous volume of data, particularly considering that the researchers - in contrast to genome analysis - do not just want to identify all proteins in the cells. To understand all of the cell's processes, they must also know the quantities in which these proteins exist and how they are further changed in the cells.
While analysis of this data is very difficult and poses particular bioinformatic challenges, it also offers the opportunity to investigate diseases in a much better manner than was previously possible. After all, proteins are indispensable building blocks of life, without which a cell cannot survive. These gigantic molecules composed of amino acid chains play a role in the development of many diseases, such as cancer. Proteins changed by diseases can lead to uncontrollable growth of cells, just like missing proteins can, or those formed at the wrong time in the wrong place.
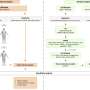
Matthias Mann and his team are developing methods that enables them to identify many proteins at once. The physicist and mathematician Mann took up the position of Head of the department of Proteomics and Signal Transduction at the Institute in Martinsried six years ago. The Max Planck researchers break down proteins into smaller pieces called peptides and use a so-called mass spectrometer to ionise them and sort them according to their masses in an electrical field. Based on the distribution and strength of the peptide signals and their fragments in the measuring instruments, the researchers can reconstruct the proteins and even their quantities.

To be able to do this, however, the scientists must first know what they are looking for. To date, only relatively few proteins are known that are indicative of various cancer types. This is where Tami Geiger comes in: over the past three years, the young Israeli scientist has been setting up a protein database of key cancer cell lines. She uses this library as a reference in comparing human tissue samples from patients. To compare the samples of a healthy person with those of a cancer patient, for example, she labels proteins with specific carbon or nitrogen isotopes which are heavier than naturally occurring atoms. On the basis of the signal strength of the marked and unmarked peptides in the mass spectrometer, Tami Geiger can recognise whether a protein is formed more strongly or more weakly within cancer cells.
Towards a complete protein catalogue with super-SILAC
With this method, known as "super SILAC", the Israeli researcher analyses the tissue samples of hundreds of breast cancer patients. Individual differences between the patients that are not related to their cancer illness are not relevant to her evaluation. "What remains are the proteins that differ in terms of healthy and degenerative cells", explains Tami Geiger. The objective is to establish an extensive catalogue of protein markers. Her colleagues are working on a catalogue for cancer of the colon and prostate.
In the not too distant future, medical researchers may be able to use this method to test if a sample contains cancer cells. "To this end, the technology must be robust, easy to use, and not too expensive. Our method already largely fulfils these requirements. But it could take many more years until it has made its way into the clinics", suspects Matthias Mann.

Thanks to Jacek Wisniewski, a further important step on the way to clinical application has already been made. The chemist has developed a method for proteome analysis that makes it possible to prepare proteins from fixed, preserved tissue samples so that they can be used for mass spectrometry. This is an important requirement for basic research because it enables scientists to examine the many tissue samples stored in pathology departments of clinics worldwide that come from patients with fully documented medical records. "In the beginning, we weren't taken seriously at all – hardly anyone believed that tissue samples that had been soaked in formalin could still yield any usable information about the proteins", says Wisniewski with a smile. But the scientist persisted and has proved all his critics wrong. Today, his method can be bought, readymade.
Proteomics vs genomics?
But what can proteomics do that genomics can't? "Gene analyses show if a certain gene variation occurs or not. In contrast to protein analysis, it doesn't tell us directly about the activity of the gene. This information, however, is incredibly important to be able to understand life processes and diseases", says Matthias Mann.
And this is not the only advantage that proteome analysis has over genome analysis: diseases are not only caused when proteins are produced in too large or too small quantities. To enable proteins to pass on signals between and within cells, various molecules are subsequently added. Phosphate additions, for example, can switch a protein on or off. Others decide where a protein is transported within a cell or if it is degraded by the cell’s built-in cleaning crew. Many diseases are caused by aberrant changes in these complicated regulatory mechanisms.
These modifications are made visible in the mass spectrometer. In 2006, Matthias Mann and colleagues identified more than 6,600 sites in 2,200 proteins where phosphates can be added. 90 percent of these phosphorylation sites were unknown at that time. Now the researchers in Martinsried want to examine how this pattern differs in cancer cells and healthy cells.
In the future, medical researchers will be able to determine how high the risk is for a cancer patient through the analysis of tumour proteins, whether tissue already contains cancerous cells and if a given tumour is likely to form metastases. It might also be possible to predict the effect of drugs in this manner – possibly even more accurately and clearly than with gene analyses. For Matthias Mann, however, both procedures are more than just two sides of a coin. "They do not mutually exclude one another and but will complement each other in the prediction and treatment of diseases".