Study sheds light on cancer-causing gene regulation
Researchers at Beth Israel Deaconess Medical Center (BIDMC) have uncovered the genes that regulate MDM2, an oncogene that, in turn, regulates the tumor suppressor protein p53. But instead of an on-off switch for MDM2, the team found what looks like a dimmer switch, suggesting a more complicated signaling pathway that is sensitive to a changing environment.
Reported in the Aug. 17 issue of the journal Cancer Cell, this new understanding of the upstream genes involved in the p53 cellular signaling pathway could point to new drug targets to help kill tumors. The discovery also points to potential biological markers for cancer risk that could one day help patients take preventive measures against cancer.
p53 guards the body against tumors and is compromised in 50 percent of cancers. Known as the guardian of the genome, p53 is part of a well-established defense system that involves a variety of proteins, and works to prevent cancer by suspending the normal cell division cycle and giving cells time to repair the dings and nicks in DNA that come from everyday environmental assaults. If the damage is too severe to be repaired, p53 triggers cell death.
Because p53 is disruptive, it exists in a "yin and yang"-like balance with its opposite, MDM2, says Wenyi Wei, PhD, Assistant Professor of Pathology at BIDMC and Harvard Medical School. "When DNA is damaged, MDM2 backs off and allows p53 to pause the cell and make repairs," explains Wei. "And when MDM2 reappears, p53 vanishes and the cell cycle rolls along normally." However, MDM2 is also known as an oncogene because an overabundance of MDM2 shuts down p53 completely, thereby compromising its cancer-preventing abilities.
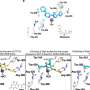
In this new study, Wei and his coauthors found that rather than simply fading away, MDM2 is actively degraded by a pair of enzymes working together. One enzyme, called Casein Kinase I (CKI), is activated when the cell detects DNA-damage, though the specifics of this activation aren't well understood. Its job is to phosphorylate MDM2, which triggers MDM2 destruction by the other enzyme, beta-TRCP1. beta-TRCP1 does this work by tagging MDM2 with a small protein called Ubiquitin, which is a little like slapping a "TRASH" tag on an unwanted piece of furniture. 26S proteasome then works as a garbage collector to clear away MDM2 and unleash the DNA-repair work of p53.
Curiously, Wei found that applying a single phosphorylation tag to MDM2 by CKI wasn't enough to guarantee that beta-TRCP1 would destroy the protein. "It's more like a dimmer switch than an on-off switch," says Wei. The team found between 17 and 23 phosphorylation sites—places on the protein that a "TRASH" tag will stick to. "It seems the cell wants to be able to adjust to the environment," says Wei. "Rather than yes or no, you have a gradual yeeeessss and then a full yes, this protein is marked for destruction."
"This new work defines how MDM2 is regulated by protein degradation after DNA damage. Although it has long been known that p53 is activated, the mechanism by which this occurs has been much less clear," said William Hahn, Associate Professor of Medicine at Harvard Medical School. "These types of experiments may eventually lead to ways to intervene in this pathway therapeutically."
Since over half of tumors have too little p53, an existing therapeutic strategy under development involves inhibiting MDM2 in tumor cells, which, in turn, will allow p53 to do its repair work or destroy cells that cannot be repaired.
One new therapeutic possibility is to move further upstream in the signaling pathway to promote CKI. Wei speculates that increased levels of CKI may reduce MDM2 and unleash p53. However, says Wei, "it is easier to make an antagonist to inhibit a protein than it is to make an agonist to encourage one."
Another possibility is that clinical researchers may want to look at clinical tumor samples to see if CKI or beta-TRCP1 are mutated in different forms of cancer. If so, these mutations could become biomarkers of cancer risk. As such, they could encourage patients to take preventive measures, similar to the way the BRCA1 gene involved in breast cancer risk may influence patients with extremely high cancer risks to undergo preventive surgery.
Wei's work, which so far has focused on studies of cell cultures and mice, will continue with further efforts to understand how DNA-damage induces the p53 repair pathway. His work, along with recent findings from co-author Galit Lahav of the HMS Department of Systems Biology, also shows that cells show a regularly pulsing ebb and flow of p53 during DNA repair that Wei would like to better understand. "It's not like the p53 levels are high or low. The dynamics in the cell are always changing," says Wei.