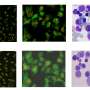
A mutation that frustrates DNA repair likely contributes to Fanconi anemia
 as part of the pathway involving other proteins such as FANCD2 (green) that rush to sites of DNA damage in cells and work together (yellow) to fix the problem.")
(PhysOrg.com) -- After more than a century of technological refinements, zippers still get stuck. So do the molecular machines that routinely unzip the double helix of DNA in our cells after billions of years of evolution, and the results can be lethal.
In research published last week in Molecular Cell, scientists at Rockefeller University and colleagues show how a previously uncharacterized protein associated with the cancer-causing disorder Fanconi anemia might aid in repairing the broken zippers in our genes. When two strands of DNA remain stuck together, they cannot replicate themselves or produce the transcripts they are supposed to make.
This sticking point is one of the most deadly genetic perils, called an inter-strand crosslink, which threatens an average cell about 10 times a day.
“Repairing the inter-strand crosslink is a very complicated process,” says Agata Smogorzewska, head of the Laboratory of Genome Maintenance at Rockefeller, who led the research. “It takes lots of players, and if they don’t work correctly, the consequences can be terrible.”
Thirteen proteins are known to be involved in the Fanconi anemia pathway, which repairs inter-strand crosslinks. If any one of them is damaged, the result is Fanconi anemia, a disorder that leads to bone marrow failure and leukemia, among other cancers, as well as many developmental abnormalities.
Seeking other proteins that may play a part in the disease, Smogorzewska and colleagues developed a test to screen thousands of genes for those that might be involved in repairing DNA damage. Using a vast library of small hairpin RNAs, which can silence genes by preventing their transcripts from being translated into proteins, the team eliminated each of more than 32,000 human proteins from cells in the test tube. The researchers bombarded the defective cells with a molecule that causes crosslinks to test whether the missing protein impaired the cells' ability to survive after the damage.
These tests winnowed the list to 2,000 genes. "Then we had to narrow it down," Smogorzewska says. "So we did additional assays, looked at the domain architecture of candidate proteins and found one that is particularly interesting." A gene, which they dubbed FAN1, had two properties that suggested it could be important to DNA repair. One region of the gene coded for what’s called a nuclease, an enzyme that cuts nucleic acids, the stuff that makes up DNA. "It also had a UBZ domain - which usually binds proteins that are embellished by ubiquitin, a very common modification found during DNA damage response generally that is essential during crosslink repair."
The team wanted to confirm FAN1's role in DNA damage response, so they tagged the gene with a fluorescent protein and studied what happened when they damaged DNA, both by using a laser to cut the DNA and a molecule that induced inter-strand crosslinks. FAN1 glommed onto sites of the DNA damage. The researchers found that when they induced a mutation in the UBZ domain of the gene, it was rendered useless, meaning its restorative ability likely lies in its capacity to interact with ubiquitinated proteins in the Fanconi anemia pathway. As predicted, the nuclease domain of FAN1 was able to cut DNA, activity that is essential repairing the broken zippers.
Versions of the gene are present in distantly related organisms, from yeast to flies to humans, Smogorzewska says, suggesting a role in life’s fundamental machinery for repairing DNA. The team’s results make FAN1 a strong candidate to add to the list of the 13 mutations currently known to disrupt the DNA damage response in Fanconi anemia patients. Because it is also associated with a group of genes that predispose women for breast cancer, the researchers recommend that FAN1 be investigated for a role in that disease as well.
More information:
Molecular Cell 39: 36-41 (July 9, 2010). A Genetic Screen Identifies FAN1, a Fanconi Anemia-Associated Nuclease Necessary for DNA Interstrand Crosslink Repair. Agata Smogorzewska, et al.
www.cell.com/molecular-cell/abstract/S1097-2765%2810%2900464-8
Provided by Rockefeller University