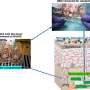
Genetic patterns rise from huge yeast samples

Princeton University scientists have developed a new way to identify the hidden genetic material responsible for complex traits, a breakthrough they believe ultimately could lead to a deeper understanding of how multiple genes interact to produce everything from blue eyes to blood pressure problems.
Writing in the April 15 edition of Nature, scientists led by Leonid Kruglyak, a professor in Princeton's Department of Ecology and Evolutionary Biology and Lewis-Sigler Institute for Integrative Genomics, report that they developed a straightforward method for studying millions of yeast cells at the same time.
Their method allows them to identify regions of the genome that cause a specific trait in the offspring of two yeast strains that have been mated. In using such a large group, the scientists have been able to identify subtle patterns that could not be detected before.
"One of the important insights gained from research enabled by the sequencing of the human genome is that, rather than being obvious, the connections between genes and most traits are very complicated," Kruglyak said. "Our results show, however, that it is possible to identify many of the factors underlying complex traits using straightforward techniques."
The Princeton team's finding could help illuminate the answers to the current difficulties inherent in tying traits to genes, known as the "missing heritability problem," Kruglyak said.
There are some cases, he said, where scientists have identified mutations in single genes that produce a specific trait, such as a susceptibility to cystic fibrosis or Huntington's disease. In most cases, however, scientists believe that large numbers of genes working in concert produce trait variation. Some genes play a major role while others are more "quiet" but still are important. Scientists want to know all of the genes involved in producing a given complex trait, but they have not been able to find these groupings, leading to the "missing" problem.
Kruglyak was part of an expert panel the National Human Genome Research Institute convened last year on the missing heritability problem. When the Human Genome Project was completed in 2003, it provided an entire sequence of human DNA. The panel discussions centered upon the idea that, despite major technology advances made possible by the project and studies of tens of thousands of individuals, the great majority of the genetic factors responsible for differences between individuals have not yet been found.
"In many cases, the effects of genes are so small that detecting them is extremely difficult," said Ian Ehrenreich, a postdoctoral research fellow who is the first author on the Nature paper. "Under conventional methods, we just don't have the power to identify many of these genes. We knew we had to find a different way."
The method described in the paper is "a creative adaptation of existing family-based, genome-wide methodology," said Yin Yao, who is chief of the molecular and genetic epidemiology program in the division of neuroscience and basic behavioral science at the National Institute of Mental Health. She and Thomas Lehner, chief of the genomic research branch at the institute, said Kruglyak's work is highly valued and described him as a pioneer in the field of statistical genomics.
Scientists in Kruglyak's lab conduct experiments on organisms like yeast, as well as perform computational analyses, aimed at understanding how changes in DNA are shaped by molecular and evolutionary forces. They also study how these changes lead to the observable differences among individuals within a species. For this research project, the team looked to develop a process that would identify genetic associations with observable traits.
 were grown at cell densities that decrease from left to right. Sensitive strains only are capable of growing when plated at high density, while resistant strains are capable of growing even at low density. By studying very large populations of yeast, Princeton scientists have developed a new way to identify the hidden genetic material responsible for genetically complex drug resistance traits. Credit: Ian Ehrenreich/Kruglyak Lab")
"We know in the human genome there are 20,000 genes, but I can't ask someone to point out to me which genes account for most of the variation in human height, for example, because we just don't know," Kruglyak said. "The underlying goal of what we are trying to do is both understand how complicated these patterns are and try to come up with some concrete examples where we can take some traits and nail down most of the variations, as opposed to only finding a small percentage."
Studies in model organisms like yeast -- just as in humans -- have failed to detect a large fraction of the genes believed to underlie most complex traits. So Kruglyak and his team developed a method using a sample size of yeast that went beyond the scale of any human studies. They crossed two strains of yeast, generating about 10 million offspring. Each of these progeny was genetically distinct as opposed to being a clone.
To find a subset offspring that shared a trait, the scientists grew the progeny on a chemical that causes breaks in DNA, killing most of them. They then sequenced the genomes of the few thousand yeast that survived, looking to see what genes they inherited from each parent.
Mendel's laws, which explain the principles of heredity, state that most of the genetic material should be randomly inherited from one or the other parent in a manner equivalent to a coin flip --half of the cells should have one parent's genes and half the other parent's. But, at the locations of the genes that protect yeast from the chemical, most of the cells should have genetic material from the parent with the version of the gene that produces greater resistance.
The surviving yeast cells' genomes were placed on tiny chips and scanned on automated laboratory machines, a process known as genotyping. The machines surveyed each yeast cell's genome for strategically selected markers of genetic variation. They found certain genetic variations to be significantly more frequent in the surviving yeast, serving as a powerful pointer to the regions of the genome where the genes underlying the trait resided.
The scientists repeated this experiment with other chemicals that were toxic to most of the yeast, looking again and again for skewed genetic patterns of inheritance. Each time, they were able to locate gene regions pointing to specific traits, confirming that the method worked.
Viewing their success, Ehrenreich said, "It's really been a combination of having the technology to do this genotyping precisely and also being able to survey such a large number of individuals simultaneously."
From here, the team intends to use the information it has on gene regions and markers to fine tune the method and identify the specific genes associated with each trait, and to extend the method to many other yeast strains.
Detlef Weigel, director of the Max Planck Institute for Developmental Biology in Germany, already sees additional applications for the technique. "The new work by Dr. Kruglyak and colleagues beautifully showcases how new sequencing technologies are revolutionizing genetics," he said. "While the work was carried out with yeast, I am convinced that it can be easily extended to any other genetically tractable organism, including crop plants."
Provided by Princeton University