Gene linked to a rare form of progressive hearing loss in males is identified
A gene associated with a rare form of progressive deafness in males has been identified by an international team of researchers funded by the National Institute on Deafness and Other Communication Disorders. The gene, PRPS1, appears to be crucial in inner ear development and maintenance. The findings are published in the Dec. 17 early online issue of the American Journal of Human Genetics.
"This discovery offers exciting therapeutic implications," said James F. Battey, Jr., M.D., Ph.D., director of the NIDCD. "Not only does it give scientists a way to develop a targeted treatment for hearing loss in boys with this disorder, it may also open doors to the treatment of other types of deafness, including some forms of acquired hearing loss."
The gene is associated with DFN2, a progressive form of deafness that primarily affects males. Boys with DFN2 begin to lose their hearing in both ears roughly between the ages of 5 and 15, and over the course of several decades will experience hearing loss that can range from severe to profound. Their mothers, who carry the defective PRPS1 gene, may experience hearing loss as well, but much later in life and in a milder form. Families with DFN2 have been identified in the United States, Great Britain, and China.
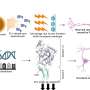
The NIDCD-funded researchers led by Xue Zhong Liu, M.D., Ph.D., of the University of Miami Miller School of Medicine, discovered that the PRPS1 gene encodes the enzyme phosphoribosylpyrophosphate (PRPP) synthetase 1, which produces and regulates PRPP (phospho-ribosylpyrophosphate), and appears to play a key role in inner ear development and maintenance. The four mutations identified in the PRPS1 gene cause a decrease in the production of the PRPP synthetase 1 protein that results in defects in sensory cells (called hair cells) in the inner ear, and eventually leads to progressive deafness.
"PRPS1 is an interesting example of a human disease gene in which gain of function or loss of function mutations cause several different and distinct hereditary disorders," says Dr. Liu. "Our findings emphasize the body's need for tight regulation of PRPP synthetase 1 since a drop in activity can lead to deafness." Other mutations in the PRPS1 gene have been linked to neurodegenerative disorders such as Arts syndrome and a form of Charcot-Marie Tooth disease, both of which feature deafness in the constellation of symptoms.
Knowing that a reduction in the amount of PRPP synthetase 1 is what causes deafness in DFN2, Liu and his colleagues are now exploring potential enzyme replacement therapies to either restore hearing or prevent further hearing loss in boys with DFN2. They believe that since the PRPS1 mutations can be used as a genetic marker for DFN2, in the future at-risk boys could be tested at birth and immediately put on enzyme replacement therapy to reduce or prevent the hearing loss that would ordinarily come later in life.
In addition, the knowledge that scientists gather about the mechanisms of PRPS1 potentially could be used to develop treatments to combat acquired hearing loss, such as the hearing loss caused by drugs that are used in some chemotherapy regimens and treatments for HIV/AIDS. These are powerful and helpful medications, but they have the unfortunate side effect of damaging, even killing, hair cells in the inner ear. The results from this study open the possibility for improving these life-saving treatments by eliminating or reducing the disabling side effect of hearing loss.