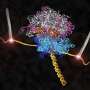
Computational microscope peers into the working ribosome (w/ Video)
 resides in the membrane. Credit: Theoretical and Computational Biophysics Group, University of Illinois Beckman Institute.")
Two new studies reveal in unprecedented detail how the ribosome interacts with other molecules to assemble new proteins and guide them toward their destination in biological cells. The studies used molecular dynamics flexible fitting (MDFF) to examine the interaction of the ribosome with two prominent molecular partners.
The first study, in Proceedings of the National Academy of Sciences, concerns the intimate signaling between the ribosome and an elongation factor (EF-Tu) that is essential to the successful assembly of a growing protein. The second, in the journal Structure, focuses on a membrane protein called SecY that sometimes latches onto the ribosome and guides a newly forming protein toward its final destination.
MDFF combines data from two reliable but limited sources of atomic-level information: X-ray crystallography, which can give a high-resolution picture of a single type of molecule, but only in a static, crystal structure; and cryo-electron microscopy (cryo-EM), which can image the dynamic, real-life interaction of two or more molecules in the cell, but at low resolution.
The problem with X-ray crystallography is that the molecules, in this case ribosomes, are artificially removed from the environment of the cell and "packed together like sardines," said University of Illinois physics professor Klaus Schulten, an author on both papers and principal investigator on the study in Structure. This gives researchers a very detailed image of the components of the ribosome, but offers no clues about its behavior when it encounters other molecules.
Cryo-EM offers a wealth of information about the ribosome in its natural habitat in the cell, but the picture is much less crisp. Like a blurry photograph of a football player maneuvering down the field, cryo-EM gives a general outline, a three-dimensional snapshot of the molecule or molecules of interest at a given point in time, Schulten said.
"It is like a cloud that gives you the volume within which you find 90 percent of all the electrons of the system," he said. The clouds capture the ribosome in action, but require computing to reveal chemical detail.
The researchers began by building computerized, atomic-scale models of the ribosome-protein complexes based on the crystal structures of the molecules, and then directed the computer to use this information to "fit" the structures into the electron clouds seen in cryo-EM studies. Simulations tracked the behavior of 2.7 million atoms in the SecY study, making it "the biggest, or one of the biggest, computer simulations to be published so far," Schulten said.
In the first study, the researchers were able to detect the precise molecular maneuvering that allows the ribosome and EF-Tu to recognize and interact with another molecule, transfer-RNA (tRNA). This interaction is key to the successful assembly of proteins because the ribosome and its partners must recognize the tRNA that carries the correct amino acid to be added to the growing protein chain.
The researchers on this study, led by Joachim Frank, of Columbia University (who also provided the cryo-EM data), found structural evidence that when the ribosome recognizes the correct tRNA it induces a change in the shape of EF-Tu. A gate in EF-Tu swings open, allowing a cascade of chemical interactions that lead to the addition of the amino acid to the protein.
The second study provided robust evidence that when the ribosome is translating a membrane protein, or a protein destined for excretion, it hooks up with a single SecY membrane channel shortly after protein translation begins.
The SecY binds to the ribosome by inserting two looped strands into the ribosome's exit channel. This interaction loosens a plug that normally seals the SecY channel. The plug moves out of the way, allowing the ribosome to funnel the growing protein through the membrane channel.
"We simulated the process of translocation of a (protein) out of the ribosome and into the SecY channel," said James (J.C.) Gumbart, a postdoctoral researcher at Illinois and first author on the Structure study. "And so we find that even though these loops are inserted into the (ribosome's) exit tunnel, they are not disturbed, nor do they get in the way of a nascent protein coming out."
Schulten directs the theoretical and computational biophysics group at the Beckman Institute for Advanced Science and Technology. He and his colleagues pioneered the MDFF approach and, thanks to support from the National Center for Research Resources at the National Institutes of Health, have made its software freely available to more than 160,000 users, he said.
Crystallographers and those doing cryo-EM are enthusiastically embracing MDFF, Schulten said, as this software can be used to tease out the elusive details of otherwise ambiguous data.
In these and other upcoming studies, Schulten and his colleagues are using the computer as a microscope to get a clearer picture of the dynamics of the ribosome, which is perhaps the cell's most essential, and most complex, molecular machine.
Source: University of Illinois at Urbana-Champaign (news : web)