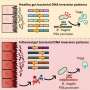
Genetic source of muscular dystrophy neutralized
 are seen here inside muscle cell nuclei (blue) from an individual with myotonic dystrophy.")
(PhysOrg.com) -- Researchers at the University of Rochester Medical Center have found a way to block the genetic flaw at the heart of a common form of muscular dystrophy. The results of the study, which were published today in the journal Science, could pave the way for new therapies that essentially reverse the symptoms of the disease.
The researchers used a synthetic molecule to break up deposits of toxic genetic material and re-establish the cellular activity that is disrupted by the disease. Because scientists believe that potentially all of the symptoms of myotonic dystrophy - the most common form of muscular dystrophy in adults - flow from this single genetic flaw, neutralizing it could potentially restore muscle function in people with the disease.
"This study establishes a proof of concept that could be followed to develop a successful treatment for myotonic dystrophy," said neurologist Charles Thornton, M.D., the senior author of the study and co-director of the University of Rochester Medical Center's Wellstone Muscular Dystrophy Cooperative Research Center. "It also demonstrates the potential to reverse established symptoms of the disease after they have developed, as opposed to simply preventing them from getting worse."
Myotonic dystrophy is a degenerative disease characterized by progressive muscle wasting and weakness. People with myotonic dystrophy have prolonged muscle tensing (myotonia) and are not able to relax certain muscles after use. The condition is particularly severe in the hand muscles and can cause a person's grip to lock making it difficult to perform rapid, repeated movements. Currently there is no medication to halt the progression of the disease.
Toxic RNA Holds Proteins Hostage
Although the genetic flaw that causes myotonic dystrophy was discovered in 1992, researchers studied the defect for many years before they had a clear understanding of the molecular events that ultimately produce the symptoms of the disease. Over time it became apparent that a central player in myotonic dystrophy was RNA, a versatile molecule that is very similar to DNA. RNA serves a vital function by relaying the genetic information from the nucleus - the protected area of the cell that houses DNA -out to the main body of the cell, where the instructions are used to build proteins. Every gene produces its own RNA, usually in multiple copies, and every RNA is a genetic blueprint of its parent gene.
The surprising aspect of myotonic dystrophy was that the genetic defect leads to production of a toxic RNA - the first example in human genetics in which RNA was cast in the role of molecular perpetrator. The errant RNA has a toxic effect because it grabs onto and holds hostage certain proteins, preventing them from carrying out their normal functions. For example, the capture of a protein called "muscleblind" causes the locking grip phenomenon that is a hallmark of the disease, a sign of faulty electrical control in muscle cells. Over time, the toxic RNA is produced in abundance and the captive proteins accumulate in deposits - or inclusions - that are visible in the cell's nucleus.
"An unexpected byproduct of research on myotonic dystrophy was that we were forced to change our ideas about the role of RNA in genetic disease," said Thornton. "Once we adjusted to this new concept, we realized that the prospects for developing treatment might be unusually good. No essential component of muscle is missing, but some important proteins are in the wrong place, stuck on the toxic RNA."
New Tools to Tackle Genetic Flaws
The Rochester team used a synthetic molecule - called an antisense morpholino oligonucleotide - that mimics a segment of the genetic code. In this case the morpholino was specifically designed to bind to the toxic RNA and neutralize its harmful effects by releasing the captured proteins. When injected into the muscle cells of mice with myotonic dystrophy the molecule found its way to the cell nucleus, broke up the deposits of toxic RNA, freed the captive muscleblind proteins, and ultimately improved the function of the muscle cells.
The researchers specifically observed a restoration of proper electrical control in the cells, which is a convenient way to monitor the condition. However, because the hostage proteins play a role in a myriad of other cellular functions, they believe that this treatment will ultimately alleviate other aspects of the disease as well.
"Based on our current understanding we would predict that by releasing the proteins held hostage, many of the symptoms of the disease may potentially be corrected by this approach," said URMC neurologist Thurman Wheeler, M.D., co-author of the study.
These genetic tools are relatively new and have provided researchers with a heretofore unprecedented ways to precisely target and manipulate genetic activity. "The current textbooks for medical students do not have chapters on antisense oligonucleotides, but this will change in the near future," said Thornton. "As compared to conventional drugs that work on proteins, antisense oligonucleotides work on RNA. They have been around for 20 years, but only recently is their full potential being realized. They provide great flexibility and they can be developed rapidly."
Source: University of Rochester Medical Center (news : web)