Scientists clarify editing error underlying genetic neurodegenerative disease
Two molecular biologists at Cold Spring Harbor Laboratory have uncovered important new details about how a gene mutation causes a cellular editing error that results in a devastating disease called pontocerebellar hypoplasia (PCH). The new findings were published online, ahead of print, on January 25th in the journal Nature Structural and Molecular Biology.
Typically striking during early childhood, PCH is characterized by the slow wasting away of certain parts of the brain, resulting in abnormal brain function and cognitive impairment characteristic of mental retardation. Although scientists have known about the gene mutation that causes the disease, they haven't been able to explain why the mutation causes a defect in an essential cellular function called RNA splicing.
RNA splicing is an essential step in the process in cell nuclei whereby instructions encoded in DNA are transcribed to RNA copies, which subsequently leave the nucleus to serve as templates for the cellular machinery to manufacture protein molecules. The RNA intermediaries (called "messengers"), in order to function properly in this role, are typically "edited" by special enzymes, which perform a procedure analogous to the editing of frames in a film, where unnecessary frames are left out of the final version.
An 'atypical' splice site
The new discovery stems from a program of research by Professor Adrian Krainer, Ph.D., and members of his lab at CSHL, to understand how cells process the information encoded in genes. For reasons that remain poorly understood, the raw or unedited RNA copy of DNA includes excess RNA segments called introns that need to be edited out in order for the RNA's message to be functional. Once the introns are removed, the remaining segments -- called exons -- are pasted together, forming a mature messenger RNA transcript.
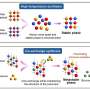
In an unedited RNA molecule, the boundaries between an intron and its two flanking exons are called splice sites. Such sites are composed of short sequences of RNA "letters" (called "bases"), which are referred to by single-letter molecular shorthands: A, U, G and C. The cell's RNA splicing machinery is correctly guided to the splice site at the beginning of an intron by one of its components, a small nuclear RNA called U1.
U1 recognizes this splice site by lining up against the target RNA and pairing a segment of its own RNA bases with the splice site's RNA bases following a set of rules: U pairs with A or G, and C pairs with G. U1's ability to recognize this splice site at the start of the intron is strongest when up to 11 bases are paired up.
When Krainer and postdoctoral researcher Xavier Roca, Ph.D., analyzed SpliceRack, a comprehensive database of all ~200,000 known, functional splice sites found at the beginning of every intron in human genes, they were surprised to find many of these sites that didn't appear to have the right sequence of RNA bases to match U1. Experimental testing of particular examples of these sites showed, however, that they were in fact recognized by U1 and effectively used. These "atypical" sites, in other words, could be spliced to make the correct messenger RNA, despite the apparent mismatch.
Atypical sites are recognized due to a shift in base-pairing
This raises the question: how does U1 manage to recognize such a diverse set of sequences, which include sequences that seem to be very poor matches to U1? The answer, Krainer and Roca have found, is explained in at least some cases by how U1 lines up with its target RNA.
"In the three decades since RNA splicing was discovered, scientists have believed that a functional match between U1 and its target RNA occurs only when the RNA bases in U1 and the target line up in a unique way," explains Roca. "We now find that this rule sometimes has exceptions."
The CSHL team has now demonstrated that U1 is more flexible in its binding than previously thought. Instead of only lining up in a manner such that a particular base in U1 pairs with the first base of the target intron's RNA sequence - that is, in the conventional way - the scientists have found that U1 can shift itself down to the next base in the intron's RNA sequence if this new arrangement allows more of U1's RNA bases to pair up with bases in the intron, thus producing a stronger match.
This phenomenon, which Krainer calls "shifting of the base-pairing," can be explained in terms of locks and keys. If the many different splice sites are thought of as different locks, then U1 is the master key that can open them all. Some locks don't match well with the master key at first try. But it has now been shown that if the key is shifted a bit so that more of the individual "serrations" of the "key" match those of the "locks," the key will work perfectly well.
Implications for Disease
This ability of U1 to slide down just a single RNA base to recognize an atypical splice site might seem like a slight adjustment, but disrupting this phenomenon can have pathological consequences.
In the specific context of the gene mutation implicated in PCH, base-pair shifting explains why this mutation causes a severe disease. Krainer and Roca's results indicate that the correct splice site of this gene is normally recognized by the sliding mechanism and not in the conventional way, as scientists had previously believed. The mutation disrupts this recognition, resulting in an abnormally "edited" RNA molecule. The "message" from the gene that is carried by the wrongly edited RNA, therefore, is faulty. This can result, in young children, in the onset of PCH.
The CSHL team's findings also have implications for studies aimed at uncovering and characterizing new disease-causing mutations, according to Krainer. "We expect to more accurately identify and understand certain splicing mutations that we may have previously overlooked," he explains.
Source: Cold Spring Harbor Laboratory