August 6, 2007 feature
Divide-and-conquer strategy key to fast protein folding

Researchers have found that proteins may use a divide-and-conquer strategy to fold into their native states in mere microseconds. The physical strategy, called “zipping and assembly” (ZA), can increase the speed at which supercomputers predict protein folding structures, greatly increasing scientists’ understanding of these building blocks of life.
The scientists, Banu Ozkan, Albert Wu, John Chodera, and Ken Dill from the University of California at San Francisco, have published their research in a recent issue of the Proceedings of the National Academy of Sciences. Their results show that the ZA search strategy provides a physics-based model of protein folding that could lead to advances such as computer-based drug discovery and genetic engineering.
“Our research has two significant points, I believe,” Dill told PhysOrg.com. “First, it shows that all-atom physical force fields are pretty good (but not perfect), and may be useful for protein structure prediction. And second, it proves that zipping and assembly is a highly efficient conformational search method, and supports the view that ZA may be the physical mechanism of protein folding.”
Proteins, which consist of an unstructured linear chain of amino acids, can fold into complex 3D structures within microseconds. On the other hand, high-speed supercomputers might take tens of years to compute the correct structure due to the vast assortment of possible forms the protein could take. When folded incorrectly, proteins can cause neurodegenerative diseases such as Alzheimer’s and mad cow disease.
How proteins fold so quickly is a mystery that researchers are approaching from many different angles, including, for example, physics-based force fields. By assigning force fields to different parts of the protein, computers can track the movement of each individual part.
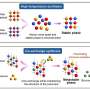
Using the ZA strategy, Ozkan, Wu, Chodera and Dill have sped up the rate at which computers using force fields can predict protein structures. In the ZA model, the first step that proteins (or computers) take is breaking the amino acid chain into 8-12 fragments to search for a very small fraction of favorable folding points (traditional methods usually search the entire chain).
“The speed gain in our method comes from not exploring all possible folding routes, but instead from following only those routes that entail small conformational search steps,” Dill explained. “This reduces the search problem, in principle, from one that grows exponentially with the chain length to one that, instead, grows only as the first or second power of the chain length. While we haven't actually proven that quantitatively, it is clear that the method is much faster than brute force Monte Carlo or molecular dynamics.”
For the fragments that contain stable hydrophobic contacts (which cause the protein to fold), the protein/computer enforces these contacts and adds more residues to enlarge these points. If new hydrophobic contacts are formed again, the process continues until no more contacts are found.
“The protein doesn't ‘know’ it has the right starting points at the early stages,” Dill explained. “It explores many possible avenues. But, we find that when the chain pieces have reached roughly the 16-to-24-mer stage, then the differences in free energy begin to become compelling, and structures begin to emerge fairly clearly.”
In some cases, this zipping procedure alone is enough for a protein to reach its native form. For other cases, the ZA algorithm switches to the assembly procedure, combining two or more fragments to form additional structures until the native form is achieved.
The ZA method was tested on nine small proteins, eight of which closely matched the experimental results of samples in the Protein Data Bank. The greatest sign that the ZA method is on the right track is its speed. For example, using ZA, protein G could fold in about 1 CPU year on a 2.8-Ghz Xeon Intel machine.
“We don't know exactly what the speed gain is,” Dill said. “Our largest protein studied is a 112-mer (subsequent to the PNAS paper). Currently the most extensive all-atom physical simulations on Folding@Home, with 100,000 processors, or IBM's Blue Gene, built for this problem, are on smaller proteins, typically in the range of 20-40 amino acids long. However, those simulations are also more directed at physical questions of folding, rather than at protein structure prediction, so it's hard to make a direct comparison.”
Citation: Ozkan, S. Banu, Wu, G. Albert, Chodera, John D., and Dill, Ken A. “Protein folding by zipping and assembly.” Proceedings of the National Academy of Sciences, July 17, 2007, vol. 104, no. 29, 11987-11992.
Copyright 2007 PhysOrg.com.
All rights reserved. This material may not be published, broadcast, rewritten or redistributed in whole or part without the express written permission of PhysOrg.com.