Closing In On Quantum Chemistry
Researchers in the Department of Energy's Lawrence Berkeley National Laboratory and the University of California at Berkeley have simulated the process by which a quantum computer could calculate to high precision an important basic property of two small molecules. Simulated quantum calculations of the ground-state energies of water (H2O) and lithium hydride (LiH) are the first of this kind ever done for specific molecules.
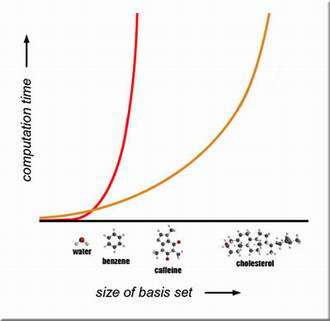
Image: An impressionistic view of why quantum computers are needed for exact solutions of the electronic structures of large molecules: as basis sets (the information needed to describe the orbital states of each particle in the system) grow, the calculation time increases, exponentially in the case of classical computers (red) but only polynomially in the case of quantum computers (orange).
Alán Aspuru-Guzik, Anthony Dutoi, Peter Love, and Martin Head-Gordon report on their work in the 9 September issue of the journal Science. Head-Gordon is a staff scientist in Berkeley Lab's Chemical Sciences Division and a professor of chemistry at UC Berkeley; Aspuru-Guzik is a postdoctoral fellow and Dutoi a graduate student in the Head-Gordon group. Love is a senior applications scientist on the staff of D-Wave Systems, Inc. in Vancouver, B.C.
The researchers developed a quantum-computational algorithm and ran it on a classical computer to demonstrate that quantum computers comprised of only tens or a few hundreds of quantum bits (qubits) could calculate significant information about real molecular systems to high accuracy. Thus a relatively small quantum computer could surpass the most powerful quantum-chemistry calculations possible with today's classical supercomputers.
"What we have done is demonstrate — by using a quantum algorithm to determine the states of minimum energy for two real molecules — that quantum computing can deliver on the promise of giving highly accurate practical solutions to interesting chemical problems," says Aspuru-Guzik.
Confronting virtually unsolvable problems
The Head-Gordon group concentrates on calculating the electronic structure of molecules from first principles — that is, from a quantum-mechanical description of the states of all the particles in the system. Electronic structure calculations allow scientists to predict how molecules react with other molecules and are key to understanding and controlling their physical and chemical properties.
The practical challenge of such calculations was famously expressed by Paul Dirac in 1929, who remarked of quantum mechanics that "The underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble."
Indeed, exact solutions of the Schrödinger equation, the fundamental expression of quantum mechanics, are so complicated that classical computers are only able to exactly solve very small molecules, about the size of water, because the time needed for computation increases exponentially with size. Practical calculations on real molecules are performed using approximations such as density functional theories. These are useful and usually accurate, but nonetheless are still approximations, which can sometimes fail. As long ago as 1982 Richard Feynman suggested that an easier way to calculate a quantum system might be by using quantum computers.
Unlike classical computing, where each bit represents either a 0 or a 1 but not both at once, a quantum bit simultaneously superposes 0 and 1 and only resolves (or "collapses") to a single value when measured. While a classical computer operates serially, essentially dealing with one bit after another, a quantum computer's qubits interact to form very large computational spaces that, when measured, quickly deliver the solution to a complex problem.
Various physical systems have been used to perform quantum computations, but no one has yet built a quantum computer large enough to compete with classical computers. Hardware is only part of the challenge. Another is devising practical algorithms that can run on quantum computers; in principle these can be run — if much more slowly — on classical simulations of quantum computers, provided only a few qubits are involved.
Aspuru-Guzik calls this the Russian doll approach: "You begin with the physical system you want to describe — that's the biggest doll, with the most information. Inside that is the basic equation that describes the system. Inside that is an 'emulation' of the system using a quantum computer. And inside that is a simulation of the quantum computer on a classical computer."
The limits to computation
Classical supercomputers are hardly tiny; rather they are limited by the number of operations they can manage within a reasonable time. Only very small molecular systems have been solved exactly from first principles, because the orbital states of each particle in the system must be represented in what's known as a basis set, which in a molecule with many electrons is very large indeed. As the size of the system increases, the number of calculations — and thus the time needed to solve the problem — increases exponentially (the larger the number gets, the faster it grows).
Using quantum algorithms on a quantum computer, however, the number of calculations (and thus the time) grows only polynomially — faster than linearly, but still "efficiently" — as the size of the basis set grows.
Aspuru-Guzik says, "We chose to calculate water and lithium hydride because three-atom water is a good-sized molecule with a small basis set, while two-atom lithium hydride is a small molecule but has a comparatively large basis set."
Two factors were key to the group's success. One was finding an efficient way to achieve the essential starting point of any calculation, an approximation of the ground-state energy sufficiently close to the actual state — the process of "preparing the state that you will simulate," as Aspuru-Guzik puts it. The researchers showed that by using a method called adiabatic state preparation (ASP), even a relatively crude initial estimate was practical.
"'Adiabatic' in this context means reiterating approximations of the state slowly," he says. "How fast you can prepare the state is determined by the gap between the ground state of the molecule and its lowest excited state. We found a way to keep this gap large." The researchers confirmed the accuracy of the ASP method by calculating the ground state of the two-electron hydrogen molecule (H 2).
Even more important was their adaptation of a quantum algorithm called a phase estimation algorithm (PEA), proposed by Daniel Abrams and Seth Lloyd six years ago. The original version required a read-out register of about 20 qubits — prohibitively large for early quantum computers. By modifying PEA so that it performed recursively, approaching greater accuracy with each repeated calculation, the researchers reduced the size of the read-out register to a manageable four qubits.
Don't try this at home
Applying these and other measures, the researchers were able to simulate a quantum computer's calculation of the ground states of water and lithium hydride with accuracy to six decimal places. A real quantum computer could have performed the same calculations almost instantly. Its classical simulacrum, however, was an order of magnitude less efficient than the best conventional methods now available, leading the authors to emphasize that "while possible as experiments, such simulations are not competitive as an alternative" to what people already do on classical computers.
"In other words, we're saying don't try this at home," says Aspuru-Guzik. "What we've done is illustrate the truth of the conjecture that to exceed the limits of classical computing, quantum algorithms running with at least 40 to 100 qubits are needed."
The members of Head-Gordon's group at Berkeley Lab and UC Berkeley continue to explore new theoretical approaches and define more specifics for the design of practical algorithms to enable quantum chemistry on quantum computers. Making the vast power and speed of quantum computers available to industrial customers is the goal of Vancouver's D-Wave Systems, Inc., the Head-Gordon group's research partner and sponsor in the work.
"Simulated quantum computation of molecular energies," by Alán Aspuru-Guzik, Anthony D. Dutoi, Peter J. Love, and Martin Head-Gordon, appears in the 9 September 2005 edition of Science.
Source: University of California at Berkeley